Análisis de componentes principales: una herramienta para dilucidar los movimientos esenciales de las biomoléculas
Principal Component Analysis: A Tool to Elucidate the Essential Movements of Biomolecules
Gustavo Pierdominici SottileJuliana PalmaEl Análisis de Componentes Principales (PCA) es un procedimiento ampliamente utilizado para examinar los datos colectados en simulaciones de biomoléculas. Su gran virtud es que permite reducir enormemente la dimensionalidad del espacio configuracional de la molécula. En criollo, esto significa que en lugar de necesitar cientos o miles de coordenadas para indicar como estan posicionados sus átomos, podemos alcanzar una muy buena descripción con solo indicar un puñado de componentes principales. Esta reducción facilita enormemente todos los análisis posteriores, ya sean cualitativos o cuantitativos. Por ello, PCA es utilizado tanto para generar animaciones de los movimientos funcionales de las biomoléculas como para calcular sus superficies de energía libre o sus entropías conformacionales.
Pero ojo: para poder aplicar PCA en forma eficaz es necesario conocer sus fundamentos teóricos, ya que ellos nos ayudan a diseñar estrategias para evitar las limitaciones del método. En este artículo, discutiremos las bases teóricas de PCA y presentaremos algunos procedimientos que permiten aprovechar al máximo las ventajas de este algoritmo.
Principal Component Analysis (PCA) is a widely used procedure for examining data collected in biomolecule simulations. Its main virtue is that it significantly reduces the dimensionality of the molecule's configurational space. In simple terms, this means that instead of needing hundreds or thousands of coordinates to indicate how its atoms are positioned, we can achieve a fairly good description by only displaying a handful of principal components. This reduction greatly facilitates all subsequent analyses, whether qualitative or quantitative. Accordingly, PCA is used both to generate animations of the functional movements of biomolecules and to calculate their free energy surfaces or conformational entropies. However, we must note that to apply PCA effectively, it is necessary to understand its theoretical foundations in order to design strategies to avoid the algorithm's limitations. In this article, we will discuss the theoretical bases of PCA and present some procedures that allow making the most of its advantages.
El Análisis de Componentes Principales (PCA, por sus siglas en inglés) es un procedimiento que se utiliza para reducir el volumen de datos utilizado para describir el estado de un sistema, tratando de minimizar la información que se pierde al hacer esta reducción [1, 2]. El algoritmo fue propuesto a principios del siglo XX [3, 4], pero inicialmente tuvo escasas aplicaciones porque resulta muy tedioso realizar “a mano” los cálculos requeridos, especialmente si el sistema considerado es grande. Pero justamente, cuando el sistema es grande, es cuando puede ser útil o necesario aplicar PCA. Posteriormente, el advenimiento de las computadoras modernas permitió que PCA sea aplicado con relativa facilidad sobre sistemas verdaderamente grandes y complejos. En la actualidad, las implementaciones de PCA son increíblemente diversas y aparecen en casi todas las ramas de la ciencia y la tecnología. Por ejemplo, PCA se utiliza en algoritmos de reconocimiento facial [5, 6], en análisis genéticos [7] y en la compresión de imágenes [8]; pero también en las ciencias médicas [9] y ambientales [10, 11], e incluso en las ciencias sociales, para revelar correlaciones entre las diferentes variables que determinan los estados de un sistema.1
Cuando se realiza una simulación de dinámica molecular de una biomolécula, se obtienen archivos enormes que contienen las coordenadas cartesianas (x, y, z) de los átomos del sistema ,2 en cada instante en el que se tomaron muestras. El problema que se plantea entonces es como transformar ese volumen descomunal de numeritos en datos inteligibles. Y en el caso particular de simulaciones de biomoléculas, estamos interesados en determinar cuáles son sus movimientos más significativos, asumiendo que los mismos son requeridos para el cumplimiento de su función biológica. Justamente en este punto, en donde la utilización de PCA resulta fundamental.
La primera aplicación de PCA para caracterizar los movimientos de las proteínas fue realizada por García en 1992 [14], quien analizó una simulación de 240 ps de Crambina y observó que los movimientos de partes distantes la proteína estaba altamente correlacionados. Esta correlación demuestra que es posible reducir la cantidad de datos requeridos para describir las fluctuaciones del sistema.3 Poco después, Berendsen y sus colaboradores propusieron utilizar PCA para transformar las coordenadas cartesianas directamente obtenidas de las simulaciones, en nuevas coordenadas con una característica muy especial: solo un puñado de ellas describe la mayor parte de las deformaciones moleculares [15].
Berendsen y sus colaboradores denominaron espacio esencial (ES por sus siglas en inglés) al subespacio descrito por este pequeño conjunto de nuevas coordenadas, y dinámica esencial (ED por sus siglas en inglés) a los movimientos que ocurren dentro de este subespacio. A partir de estos dos artículos seminales, el uso de PCA se difundió rápidamente en el análisis de las simulaciones de macromoléculas, transformándose en una herramienta fundamental al combinar simplicidad y bajo costo computacional con una gran utilidad. Entre las aplicaciones más difundidas de esta técnica podemos mencionar la determinación de los movimientos funcionales de proteínas, ADNs y ARNs [16], la caracterización de fluctuaciones de cavidades proteicas [17], la descripción de los procesos de plegamiento de proteínas [18], la identificación de los estados estables y metaestables de las macromoléculas y sus complejos [19] y los cálculos de entropía configuracional [20, 21, 22, 23, 24, 25, 26].
Para realizar PCA sobre datos colectados en simulaciones de MD, se deben tomar diversas decisiones. Y para ello, es de vital importancia conocer cómo las mismas afectarán a los resultados. ¿Qué coordenadas debemos emplear en el análisis? ¿Cuánto deberían durar las simulaciones? ¿Podemos usar una sola trayectoria, o deberíamos combinar varias? Los requisitos computacionales de PCA para una proteína globular, ¿son similares a los de una proteína desordenada o un ARN? ¿Qué podemos esperar si combinamos simulaciones de la forma holo y apo de una enzima? En las secciones siguientes, intentaremos responder a estas preguntas.
Nuestro objetivo es contribuir a que una persona sin conocimientos previos en el tema pueda tomar decisiones informadas sobre todos los detalles del procedimiento, de forma tal de estar en condiciones de utilizarlo en sus estudios o investigaciones. Asimismo, debemos señalar que existen excelentes reviews sobre las aplicaciones de PCA a simulaciones MD [27, 28, 29, 30, 31]. En ellos se discuten los temas vistos en este artículo desde otras perspectivas y con otra profundidad. Recomendamos la lectura de estos trabajos a quienes deseen adquirir una visión más amplia del tema.
1El presente texto tiene como base un review que publicamos en 2023 en la revista ChemPhysChem. El mismo se tituló On the Uses of PCA to Characterise Molecular Dynamics Simulations of Biological Macromolecules: Basics and Tips for an Effective Use [12]. Además de la traducción, para el presente trabajo realizamos una adaptación del contenido original, a fin de que su lectura resulte menos técnica y más amena, aunque ciertos tecnicismos y ciertas ecuaciones no se pueden evitar. Asimismo, hicimos agregados con aspectos que terminamos de dilucidar después de haber publicado el mencionado review. Los mismos se discuten en el artículo de la Ref. 13, que fue recientemente aceptado para su publicación.
2También se pueden obtener archivos con las velocidades de los átomos.
3Para comprender por qué ocurre esto, podemos pensar en un sistema de dos variables. Si las mismas no están correlacionadas es necesario indicar los valores de ambas para decir cómo está el sistema; pero si están perfectamente correlacionadas, sabiendo el valor de una de ellas inmediatamente conocemos el valor de la otra.
En esta sección presentaremos, en primer lugar, el procedimiento seguido para un análisis de Componentes Principales. A continuación, indicaremos lo que se consigue con cada uno de esos pasos (o sea, indicaremos cual es el resultado o la acción efectivamente realizada en cada uno). Por último, terminaremos esta sección discutiendo cuáles son las motivaciones que usualmente nos llevan a hacer PCA de simulaciones MD de biomoléculas.
3.1 El procedimientoPara presentar el procedimiento seguido en un PCA, consideraremos que el análisis se realiza sobre las coordenadas colectadas de una simulación de dinámica molecular (MD-PCA), dado que esta es la situación mas común. Por ejemplo, en simulaciones de proteínas, típicamente se emplean las coordenadas cartesianas de los carbonos alfa, o de los átomos del esqueleto proteico. Sin embargo, otras opciones son posibles y muchas veces son preferibles. Estas alternativas serán discutidas en la Sección 5.2. Asimismo, debemos señalar que un PCA también se puede realizar sobre coordenadas derivadas de otras fuentes. Por ejemplo, se puede hacer PCA de modelos moleculares generados experimentalmente, tales como la resonancia magnética nuclear (RMN) o la cristalografía de rayos X. En cualquier caso, los resultados de estos usos alternativos pueden comprenderse sin inconvenientes con base en la discusión proporcionada aquí para el MD-PCA.
Utilizaremos la letra N para indicar el número de estructuras de la macromolécula que se muestrearon de la simulación, mientras que ![]() denota a un vector que contiene las coordenadas cartesianas atómicas de la k-ésima; estructura. El n-ésimo elemento de
denota a un vector que contiene las coordenadas cartesianas atómicas de la k-ésima; estructura. El n-ésimo elemento de 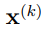 será denotado como
será denotado como 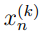 . Por lo tanto, el índice k va de 1 a N mientras que n va de 1 a 3
. Por lo tanto, el índice k va de 1 a N mientras que n va de 1 a 3 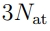 , donde
, donde 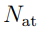 es el número de átomos incluidos en el análisis. Usualmente no incluye a todos los átomos de la macromolécula sino a un conjunto reducido. Por ejemplo, en los estudios de proteínas se suelen incluir solo los Cα o los átomos del esqueleto proteico. Discutiremos este tema con mas detalle en la Sec. 5.2. Con base en las definiciones recién elaboradas, los pasos para realizar el PCA de una macromolécula son los siguientes.
es el número de átomos incluidos en el análisis. Usualmente no incluye a todos los átomos de la macromolécula sino a un conjunto reducido. Por ejemplo, en los estudios de proteínas se suelen incluir solo los Cα o los átomos del esqueleto proteico. Discutiremos este tema con mas detalle en la Sec. 5.2. Con base en las definiciones recién elaboradas, los pasos para realizar el PCA de una macromolécula son los siguientes.
1. Eliminar del conjunto de coordenadas colectadas 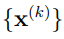 , el efecto de la traslación y la rotación global de la macromolécula. Para ello, todas las estructuras del conjunto se alinean con una de ellas, típicamente la primera.4 Si nos “salteamos” este paso (o si lo realizamos en forma deficiente) la animación del primer autovector de PCA muestra a la molécula rotando y/o trasladándose. En ese caso, los vectores que describen las deformaciones internas de la molécula (aquellas que efectivamente queremos identificar con PCA), quedan relegados a posiciones de menor importancia, lo que puede arruinar todo el análisis. Los fundamentos de los métodos de alineamiento, junto con sus detalles técnicos se discuten en la Sec. 5.1.
, el efecto de la traslación y la rotación global de la macromolécula. Para ello, todas las estructuras del conjunto se alinean con una de ellas, típicamente la primera.4 Si nos “salteamos” este paso (o si lo realizamos en forma deficiente) la animación del primer autovector de PCA muestra a la molécula rotando y/o trasladándose. En ese caso, los vectores que describen las deformaciones internas de la molécula (aquellas que efectivamente queremos identificar con PCA), quedan relegados a posiciones de menor importancia, lo que puede arruinar todo el análisis. Los fundamentos de los métodos de alineamiento, junto con sus detalles técnicos se discuten en la Sec. 5.1.
2. Calcular la matriz de covarianza C del conjunto de coordenadas . Los elementos de C están dados por,
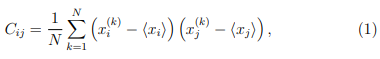
donde 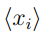 y
y 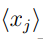 son los valores medios de las coordenadas
son los valores medios de las coordenadas 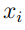 y
y 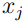 , respectivamente. Por ejemplo,
, respectivamente. Por ejemplo,

3. Diagonalizar la matriz C. Esto significa que debemos encontrar la matriz R que transforma a C en la matriz diagonal Λ mediante,

donde 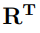 es la transpuesta de la matriz R. Dado que C es una matriz simétrica, la matriz R siempre existe, es ortogonal y puede ser transformada en ortonormal. De hecho, los programas que calculan PCA de biomoléculas siempre producen matrices R que están normalizadas. Por lo tanto,
es la transpuesta de la matriz R. Dado que C es una matriz simétrica, la matriz R siempre existe, es ortogonal y puede ser transformada en ortonormal. De hecho, los programas que calculan PCA de biomoléculas siempre producen matrices R que están normalizadas. Por lo tanto, 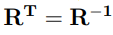 , o lo que es equivalente,
, o lo que es equivalente,

indicando que las columnas de R forman una base de vectores ortonormales (o versores). Asimismo, la gran mayoría de los programas ordenan los autovectores siguiendo un orden decreciente de sus autovalores.
Es conveniente notar que la Ec. 3 es equivalente a resolver la ecuación de autovalores,

para n= 1 . . . N , donde el vector 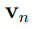 es la n-ésima columna de R, mientras que
es la n-ésima columna de R, mientras que 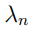 es el n-ésimo elemento diagonal de Λ. En principio, el número de autovectores con autovalores distintos de cero es
es el n-ésimo elemento diagonal de Λ. En principio, el número de autovectores con autovalores distintos de cero es 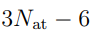 , porque este es el número de grados de libertad internos del sistema. 5Sin embargo, pueden aparecer más autovalores nulos si el tamaño de la muestra es insuficiente. Esto ocurre porque el número de direcciones requeridas para dar cuenta de las fluctuaciones de un conjunto de N puntos es
, porque este es el número de grados de libertad internos del sistema. 5Sin embargo, pueden aparecer más autovalores nulos si el tamaño de la muestra es insuficiente. Esto ocurre porque el número de direcciones requeridas para dar cuenta de las fluctuaciones de un conjunto de N puntos es 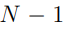 . Entonces, si realizamos PCA sobre un conjunto de N estructuras, solo obtendremos autovectores con autovalores no nulos. Aunque el sistema pueda tener otras deformaciones, estas no van a aparecer en nuestro análisis porque no le dimos al algoritmo información suficiente. En general, esta limitación no genera mayores inconvenientes porque solo un puñado de autovectores se utiliza en el análisis posterior, pero es un punto a tener en cuenta cuando se dispone de un número reducido de estructuras.
. Entonces, si realizamos PCA sobre un conjunto de N estructuras, solo obtendremos autovectores con autovalores no nulos. Aunque el sistema pueda tener otras deformaciones, estas no van a aparecer en nuestro análisis porque no le dimos al algoritmo información suficiente. En general, esta limitación no genera mayores inconvenientes porque solo un puñado de autovectores se utiliza en el análisis posterior, pero es un punto a tener en cuenta cuando se dispone de un número reducido de estructuras.
4. Definir el espacio esencial (ES) de la molécula. Para comprender lo que esto significa es necesario considerar el espectro de autovalores típico del PCA de una molécula biológica. La Fig. 1 muestra un ejemplo de estos espectros. La línea violeta de este grafico muestra los autovalores individuales en función de su índice. Se puede apreciar que la curva tiene una caída inicial muy abrupta, de manera tal que para índices mayores a 10, los autovalores son casi despreciables. Esta es una característica muy notable si consideramos que el sistema tiene un total de 354 autovalores no-nulos. El ES de la molécula se define como el conjunto formado por los primeros 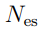 autovectores, siendo
autovectores, siendo 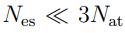 . En la Sec. 5.3 discutiremos los criterios utilizados para decidir el valor de .
. En la Sec. 5.3 discutiremos los criterios utilizados para decidir el valor de .
5. Proyectar los vectores desplazamiento, 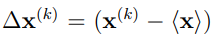 , sobre los autovectores del ES. De esta manera se obtienen los Componentes Principales (PC) para cada tiempo k en el cual se tomó una muestra,
, sobre los autovectores del ES. De esta manera se obtienen los Componentes Principales (PC) para cada tiempo k en el cual se tomó una muestra,
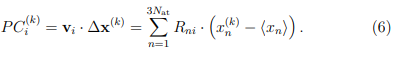
Los PCs son las nuevas coordenadas que describen la dinámica de la macromolécula en el ES. Usualmente, todo el análisis subsiguiente se realiza sobre estas coordenadas y el resto se descarta. En cierto sentido, este procedimiento implica perder parte de la información contenida en el conjunto de coordenadas . No obstante, esta acción se justifica por considerar que los movimientos que quedan afuera del ES solo aportan ruido térmico y por lo tanto complican en análisis de los movimientos relevantes.
La sección anterior describe como realizar PCA sobre la trayectoria de una macromolécula, pero no explica nada acerca de cuáles son los resultados de esos procedimientos. En otras palabras, ¿qué interpretación podemos dar a los autovectores, autovalores y Componentes Principales obtenidos con el procedimiento descrito? Esta sección está dedicada a discutir esta cuestión, a fin de que se comprendan las motivaciones para realizar PCA y se puedan estimar sus limitaciones.
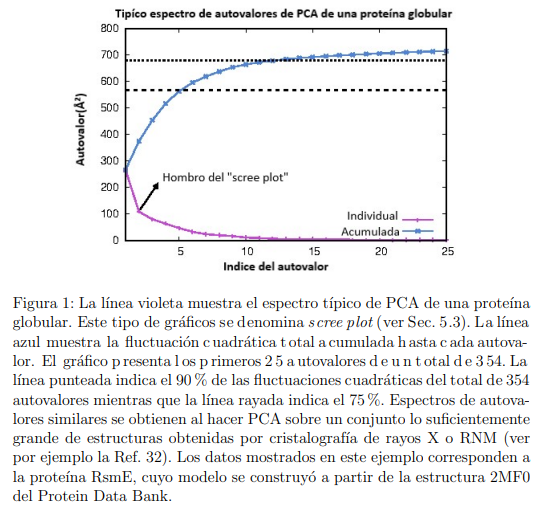
Una forma conveniente de iniciar la discusión es señalar que la conformación de una biomolécula puede ser proporcionada de maneras alternativas. Una de ellas, quizás la más común, es usar el vector 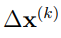 , cuyas componentes miden los desplazamientos cartesianos de los ´atomos desde su posición promedio
, cuyas componentes miden los desplazamientos cartesianos de los ´atomos desde su posición promedio
Alternativamente, podríamos emplear el vector 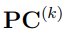 , cuyos elementos son los Componentes Principales. Estas dos representaciones están relacionadas por cambio en la base utilizada para expresar al vector que señala la configuración del sistema dentro de s u e norme espacio de configuraciones. Veamos, con un ejemplo sencillo, qué queremos decir con esto.
, cuyos elementos son los Componentes Principales. Estas dos representaciones están relacionadas por cambio en la base utilizada para expresar al vector que señala la configuración del sistema dentro de s u e norme espacio de configuraciones. Veamos, con un ejemplo sencillo, qué queremos decir con esto.
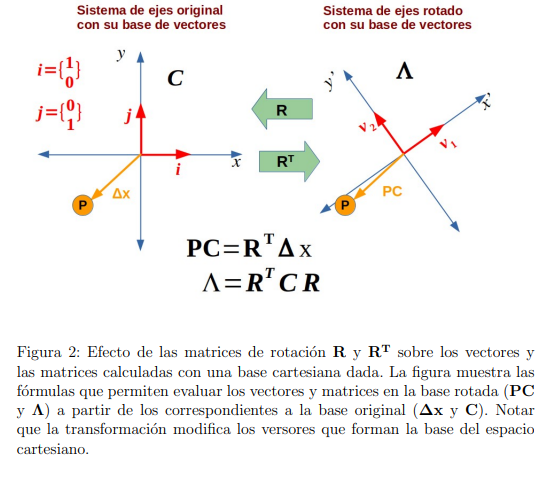
La Fig. 2 muestra un cambio de base muy simple. Corresponde a una rotación de los ejes coordenados en 2D (o sea, en un plano). Los vectores ∆x y PC tienen el mismo origen y ambos señalan la ubicación del punto P . Por lo tanto, son el mismo vector. Sin embargo, los llamamos de manera diferente porque están expresados en dos bases distintas. Para el vector ∆x utilizamos la base original 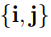 , mientras que para PC usamos la base rotada
, mientras que para PC usamos la base rotada  . Al estar expresados en diferentes bases, los componentes del vector cambian, a pesar de señalar el mismo punto. Del mismo modo, las matrices C y Λ son una misma matriz expresada en dos bases alternativas. Eso significa que Λ es la matriz de covarianza en la base transformada, o sea, es la covarianza evaluada a partir de las fluctuaciones de los PCs. Por lo tanto, los elementos diagonales de Λ miden las fluctuaciones cuadráticas promedio de los PCs; y el hecho de que Λ sea diagonal nos indica que los PCs no están correlacionados entre sí.
. Al estar expresados en diferentes bases, los componentes del vector cambian, a pesar de señalar el mismo punto. Del mismo modo, las matrices C y Λ son una misma matriz expresada en dos bases alternativas. Eso significa que Λ es la matriz de covarianza en la base transformada, o sea, es la covarianza evaluada a partir de las fluctuaciones de los PCs. Por lo tanto, los elementos diagonales de Λ miden las fluctuaciones cuadráticas promedio de los PCs; y el hecho de que Λ sea diagonal nos indica que los PCs no están correlacionados entre sí.
La única diferencia entre el ejemplo de la Fig. 2 y la transformación de la Ec. 3 es la dimensión de los vectores y matrices involucrados. Cuando indicamos la configuración de una molécula mediante un vector 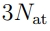 cuyos elementos están formados por ceros en todas las posiciones excepto en una, en la que hay un 1.6 Luego, las componentes de , estamos utilizando como base los autovectores de la matriz de covarianza (Ec. 5). Por tanto, para obtener los componentes del vector (o sea los
cuyos elementos están formados por ceros en todas las posiciones excepto en una, en la que hay un 1.6 Luego, las componentes de , estamos utilizando como base los autovectores de la matriz de covarianza (Ec. 5). Por tanto, para obtener los componentes del vector (o sea los 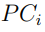 en la muestra k) tenemos que proyectar sobre esta nueva base, como indica la Ec. 6.
en la muestra k) tenemos que proyectar sobre esta nueva base, como indica la Ec. 6.
El procedimiento empleado para hacer PCA tiene muchas similitudes con el análisis de modos normales (NMA por sus siglas en inglés) [33], que se estudia en las carreras de Física, Química y Bioquímica. Discutiremos aquí estas analogías, ya que la discusión puede ayudar a quienes estén familiarizados con
NMA a comprender lo que implica hacer PCA. En el análisis de modos normales, la configuración de una molécula es inicialmente indicada por el vector 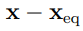 de dimensión , cuyas componentes miden el desplazamiento de cada coordenada atómica cartesiana desde su posición de equilibrio. Sin embargo, cuando la energía potencial de la molécula se expande hasta segundo orden en estas coordenadas, generalmente aparecen términos cruzados que no son despreciables. Esto implica que la matriz Hessiana, H, no es diagonal, razón por la cual los movimientos de las diferentes coordenadas están acoplados. Debido a que este acoplamiento dificulta el análisis posterior, uno busca realizar un cambio de coordenadas que convierte al vector en el vector de modos normales q, de manera tal que H es diagonal cuando se calcula con los componentes q [34].
de dimensión , cuyas componentes miden el desplazamiento de cada coordenada atómica cartesiana desde su posición de equilibrio. Sin embargo, cuando la energía potencial de la molécula se expande hasta segundo orden en estas coordenadas, generalmente aparecen términos cruzados que no son despreciables. Esto implica que la matriz Hessiana, H, no es diagonal, razón por la cual los movimientos de las diferentes coordenadas están acoplados. Debido a que este acoplamiento dificulta el análisis posterior, uno busca realizar un cambio de coordenadas que convierte al vector en el vector de modos normales q, de manera tal que H es diagonal cuando se calcula con los componentes q [34].
El paralelismo que existe entre PCA y NMA podría llevarnos a pensar que hacemos PCA simplemente para encontrar nuevas coordenadas que tengan covarianza nula7, así como hacemos NMA para que el Hessiano no tenga términos cruzados. Sin embargo, no es este el caso. La falta de covarianza o correlación en las variables generadas por PCA es simplemente un factor concurrente, no el objetivo principal. Para identificar ese objetivo, es necesario considerar la manera en la cual fueron derivadas las ecuaciones 3 y 5, que constituyen la esencia del algoritmo PCA. Esto puede hacerse de diferentes maneras [35, 36], pero la más esclarecedora requiere definir la fluctuación cuadrática media total (TMSF) de la molécula,

En esta expresión, 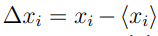 , mide el desplazamiento instantáneo de la coordenada respecto a su posición promedio, , mientras que los bra-kets
, mide el desplazamiento instantáneo de la coordenada respecto a su posición promedio, , mientras que los bra-kets
nos indican que debemos calcular el promedio de 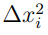 en toda la trayectoria. Por lo tanto, TMSF es la suma de los promedios de las fluctuaciones cuadráticas de todas las coordenadas del sistema. Cuanto menos estructurada y más laxa es la molécula, mayor es su TMSF. Típicamente, además, todas las coordenadas contribuyen a TMSF de forma más o menos similar, aunque hay regiones más móviles que otras y estas son las que contribuyen mayor medida.
en toda la trayectoria. Por lo tanto, TMSF es la suma de los promedios de las fluctuaciones cuadráticas de todas las coordenadas del sistema. Cuanto menos estructurada y más laxa es la molécula, mayor es su TMSF. Típicamente, además, todas las coordenadas contribuyen a TMSF de forma más o menos similar, aunque hay regiones más móviles que otras y estas son las que contribuyen mayor medida.
Cuando hacemos PCA, buscamos determinar una transformación de coordenadas tal que, en el nuevo sistema, el TMSF se concentre en unas pocas coordenadas en lugar de estar equitativamente repartido entre todas ellas.8 Con este fin, se plantea la siguiente pregunta: ¿cuál es la combinación lineal de desplazamientos cartesianos que da cuenta de la mayor fracción de TMSF? Contestar esta pregunta requiere buscar conjunto de coeficientes 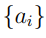 tales que la nueva coordenada s, definida como,
tales que la nueva coordenada s, definida como,
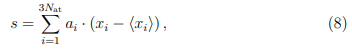
tenga la mayor fluctuación cuadrática posible, 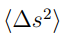 . Si además requerimos que los coeficientes estén normalizados
. Si además requerimos que los coeficientes estén normalizados 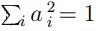 , puede demostrarse que este problema de optimización conduce a la Ec. 5. Por lo tanto, los coeficientes
, puede demostrarse que este problema de optimización conduce a la Ec. 5. Por lo tanto, los coeficientes 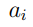 que estamos buscando son los elementos del vector , una de las columnas de la matriz R. Sin embargo, ellos no son cualquier columna de R, sino la que corresponde al mayor autovalor. Por convención, esta columna se ubica en el primer lugar de manera tal que
que estamos buscando son los elementos del vector , una de las columnas de la matriz R. Sin embargo, ellos no son cualquier columna de R, sino la que corresponde al mayor autovalor. Por convención, esta columna se ubica en el primer lugar de manera tal que 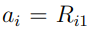 . A continuación, uno podría preguntar, ¿cuál es la combinación lineal de desplazamientos cartesianos que da cuenta de la mayor fracción de los desplazamientos cuadráticos totales remanentes, y que además es ortogonal a s. Y nuevamente, luego de requerir que los coeficientes estén normalizados, uno llega a la Ec. 5 . Los coeficientes de esta nueva variable son los elementos de la columna de R que corresponde al segundo mayor autovalor, por lo cual están ubicados en la segunda columna de R. De la misma manera, la búsqueda continúa hasta determinar las 3Nat soluciones independientes de la ecuación de autovalores. En conclusión, podemos decir que el algoritmo de PCA produce nuevas coordenadas ortogonales, diseñadas para que una de ellas sea la combinación lineal de coordenadas cartesianas con la mayor fluctuación cuadrática promedio posible, otra sea la combinación con la segunda mayor fluctuación cuadrática promedio posible y sea ortogonal a la primera, y así siguiendo. Para finalizar, es importante notar que este procedimiento determina la dirección y el tamaño de los autovectores, pero no su sentido. Por lo tanto, si se obtiene como solución un vector , debemos tener en cuenta que el vector es una opción igualmente válida.
. A continuación, uno podría preguntar, ¿cuál es la combinación lineal de desplazamientos cartesianos que da cuenta de la mayor fracción de los desplazamientos cuadráticos totales remanentes, y que además es ortogonal a s. Y nuevamente, luego de requerir que los coeficientes estén normalizados, uno llega a la Ec. 5 . Los coeficientes de esta nueva variable son los elementos de la columna de R que corresponde al segundo mayor autovalor, por lo cual están ubicados en la segunda columna de R. De la misma manera, la búsqueda continúa hasta determinar las 3Nat soluciones independientes de la ecuación de autovalores. En conclusión, podemos decir que el algoritmo de PCA produce nuevas coordenadas ortogonales, diseñadas para que una de ellas sea la combinación lineal de coordenadas cartesianas con la mayor fluctuación cuadrática promedio posible, otra sea la combinación con la segunda mayor fluctuación cuadrática promedio posible y sea ortogonal a la primera, y así siguiendo. Para finalizar, es importante notar que este procedimiento determina la dirección y el tamaño de los autovectores, pero no su sentido. Por lo tanto, si se obtiene como solución un vector , debemos tener en cuenta que el vector es una opción igualmente válida.
Ahora nos podríamos preguntar, ¿por qué es útil encontrar estas coordenadas cuando estamos analizando una simulación MD de una proteína (o de cualquier macromolécula biológica)? Para responder a esta pregunta, tenemos que pensar en los movimientos atómicos. Estos se llevan a cabo utilizando la energía térmica del sistema, que se distribuye aleatoriamente entre todas sus partículas. Sin embargo, en una macromolécula, las interacciones entre los átomos no dejan que estos se muevan de cualquier manera, sino que tienen que moverse de manera aproximadamente concertada. De esta forma, la estructura molecular y las interacciones entre sus diferentes componentes, logran canalizar la energía térmica aparentemente aleatoria en los desplazamientos colectivos que son necesarios para que la molécula pueda cumplir su función biológica. El hecho de que las fluctuaciones atómicas en las proteínas estén fuertemente correlacionadas entre sí hace que sus espectros de autovalores de PCA tengan la forma indicada en la Fig. 1. La rápida disminución inicial demuestra que unas pocas coordenadas colectivas pueden explicar la mayoría de las fluctuaciones observadas. Si los desplazamientos atómicos fueran completamente aleatorios, los espectros de autovalores de PCA solo mostrarían una disminución leve (ver, por ejemplo, la Fig. 1 de la Ref. 27).
Se suele afirmar que las coordenadas en el espacio esencial describen los movimientos funcionales de la proteína. Sin embargo, esto no es necesariamente cierto. Si las configuraciones utilizadas para l levar a cabo e l PCA corresponden al mismo pozo de energía libre, los vectores del espacio esencial describen deformaciones a lo largo de las cuales la estructura molecular es flexible. En otras palabras, la estructura molecular es propensa a ser modificada a lo largo de esas coordenadas. Esto explica los ejemplos, acumulados a lo largo de los años, que encontraron que los movimientos funcionales de las proteínas están contenidos en su espacio esencial. Sin embargo, si las configuraciones muestreadas pertenecen a dos o más pozos alternativos, la afirmación deja de ser válida. En tales casos, la conformación promedio x, que es el origen de los vectores 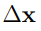 y PC, no tiene significado físico ya que el sistema nunca pasa cerca de esa estructura.
y PC, no tiene significado físico ya que el sistema nunca pasa cerca de esa estructura.
4Aunque en general el alineamiento se hace sobre la primera estructura, cualquiera de las estructuras muestreadas puede ser utilizada para tal fin.
5De las seis coordenadas restantes, tres describen la rotación global y tres la translación global de la molécula.
6Los elementos de esta base son el equivalente multidimensional de los versores bidi- mensionales i y j de la Fig. 2.
7O sea, que no estén correlacionadas.
8Además queremos que la transformación sea lineal. Pero ese es un detalle técnico.
En las secciones precedentes hemos presentado el procedimiento para hacer PCA. Además, explicamos lo que se logra con cada uno de sus pasos discutimos cuáles son las motivaciones para llevar a cabo dicho procedimiento. Muñidos de esta información, estamos ahora en condiciones de ver algunos “truquitos” que permiten sacar un mayor provecho de la aplicación de este algoritmo.
PCA de trayectorias combinadasHasta este punto, hemos considerado que el PCA se realiza sobre estructuras muestreadas de una única simulación MD. Sin embargo, una práctica muy extendida consiste en utilizar una trayectoria artificial obtenida al combinar simulaciones que emplean diferentes estados iniciales del mismo sistema. A este procedimiento también se lo llama “concatenación” de trayectorias porque los archivos de las mismas se ponen uno a continuación del otro. Las trayectorias combinadas podrían corresponder, por ejemplo, a las formas abierta y cerrada de un receptor, o a una enzima unida a diferentes sustratos, o una misma proteína en diferentes solventes. Este procedimiento fue propuesto en 1995 por Berendsen y colaboradores, quienes analizaron las diferencias entre las simulaciones de Termolisina en vacío y en agua. Desde entonces, el procedimiento se ha implementado en muchas situaciones. En la bibliografía, se conoce como Comb-ED por Combined Essential Dynamics, pero también aparece como PCA de trayectorias concatenadas. En su artículo original, Berendsen y colaboradores explicaron cómo interpretar los resultados del PCA de trayectorias concatenadas utilizando argumentos intuitivos. Mucho más recientemente, los autores de este artículo encontramos las expresiones analíticas que relacionan la matriz de covarianza de una trayectoria combinada con aquellas de las trayectorias individuales. Estas expresiones, proporcionan una base más sólida para comprender la información que proveen los componentes principales obtenidos por este procedimiento.
La matriz de covarianza de una trayectoria que combina n simulaciones
individuales de la misma molécula, 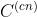 , puede expresarse en funcion de las matrices de covarianza individuales,
, puede expresarse en funcion de las matrices de covarianza individuales, 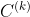 , como [37],
, como [37],
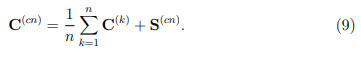
Aquí, 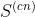 es la matriz de covarianza formada por las estructuras promedio de las simulaciones individuales. Sus elementos están dados por,
es la matriz de covarianza formada por las estructuras promedio de las simulaciones individuales. Sus elementos están dados por,
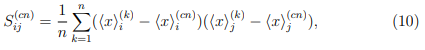
donde 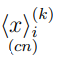 es el promedio de la coordenada i en la simulación k, mientras que
es el promedio de la coordenada i en la simulación k, mientras que 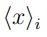 es el promedio de la coordenada i en la trayectoria combinada.
es el promedio de la coordenada i en la trayectoria combinada.
En palabras, la Ec. 9 nos dice que la matriz de covarianza de la trayectoria combinada se obtiene al sumar el promedio de las matrices de covarianza de las trayectorias individuales y la matriz de covarianza de sus estructuras promedio. Debemos observar que la matriz tiene solo n 1 autovectores con autovalores no nulos. Estos abarcan un espacio vectorial que contiene a las estructuras promedio de las simulaciones individuales.
Una forma sencilla de discutir qué se puede esperar de un PCA combinado es considerar un ejemplo particular. El más simple es el de concatenar dos simulaciones. De acuerdo con la Ec. 9, los elementos de la matriz de covarianza combinada para este caso particular están dados por:,
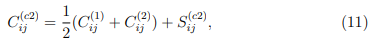
donde 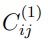 y
y 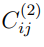 son los elementos de las matrices de covarianza calculadas con las simulaciones 1 y 2, respectivamente, mientras que
son los elementos de las matrices de covarianza calculadas con las simulaciones 1 y 2, respectivamente, mientras que 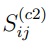 es la matriz de covarianza de sus estructuras promedio,
es la matriz de covarianza de sus estructuras promedio,
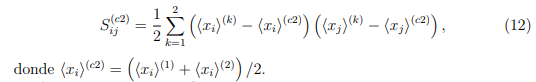
donde 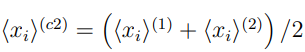 .
.
Como se discute en la Sección 3.2, los principales autovectores de las matrices 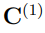 y
y 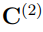 indican las direcciones de mayor fluctuación observada en cada una de las respectivas trayectorias mientras que sus autovalores miden la amplitud de esas fluctuaciones. La matriz
indican las direcciones de mayor fluctuación observada en cada una de las respectivas trayectorias mientras que sus autovalores miden la amplitud de esas fluctuaciones. La matriz 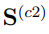 , por otro lado, se calcula a partir de solo dos estructuras. Por lo tanto, hay una sola dirección de deformación: la que nos conduce de una estructura promedio a la otra pasando por el promedio global. Esta es la dirección del único autovector de con autovalor distinto de cero. Además, como se demostró en la Ref. [37], este autovalor está relacionado con la desviación cuadrática media entre las dos estructuras promedio de acuerdo con,
, por otro lado, se calcula a partir de solo dos estructuras. Por lo tanto, hay una sola dirección de deformación: la que nos conduce de una estructura promedio a la otra pasando por el promedio global. Esta es la dirección del único autovector de con autovalor distinto de cero. Además, como se demostró en la Ref. [37], este autovalor está relacionado con la desviación cuadrática media entre las dos estructuras promedio de acuerdo con,
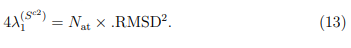
Teniendo en mente las consideraciones previas, se pueden prever dos casos límite para la diagonalización de la matriz 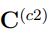 . Una posibilidad es que las dos simulaciones independientes correspondan a estructuras relativamente rígidas que muestrean regiones muy dispares del espacio configuracional. En ese caso, la distancia entre sus estructuras promedio es mucho mayor que las fluctuaciones o observadas en las trayectorias individuales. En consecuencia, la matriz hace la contribución dominante a en la Ec. 11. Si esta contribución supera ampliamente la de las matrices de covarianza individuales, el primer autovector de es casi paralelo al de . Eso implica que está prácticamente alineado con la recta que pasa por las dos estructuras promedio.
. Una posibilidad es que las dos simulaciones independientes correspondan a estructuras relativamente rígidas que muestrean regiones muy dispares del espacio configuracional. En ese caso, la distancia entre sus estructuras promedio es mucho mayor que las fluctuaciones o observadas en las trayectorias individuales. En consecuencia, la matriz hace la contribución dominante a en la Ec. 11. Si esta contribución supera ampliamente la de las matrices de covarianza individuales, el primer autovector de es casi paralelo al de . Eso implica que está prácticamente alineado con la recta que pasa por las dos estructuras promedio.
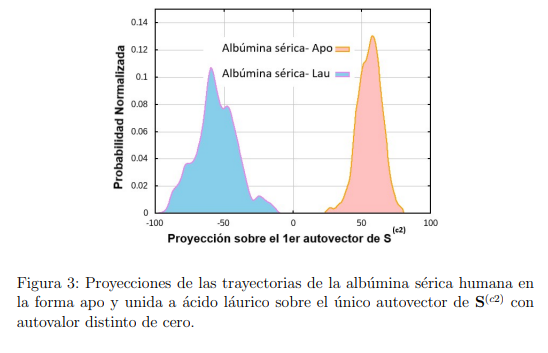
Un ejemplo de esta situación se presenta en la Fig. 3, que utiliza datos tomados de la Ref. 37 para mostrar la proyección de dos trayectorias de la albumina sérica humana sobre el primer vector obtenido al hacer un PCA combinado con ambas trayectorias. Una de las simulaciones fue realizada con la enzima unida al ácido láurico mientras que en la otra utilizó la forma apo. En la figura se puede apreciar que la distancia entre las estructuras promedio es mayor que las fluctuaciones de cada trayectoria individual. Para este ejemplo, el primer autovalor de la matriz es 4499.40 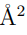 , mientras que el único autovalor de la matriz es 4361.59 . El producto escalar entre los dos autovectores se vale 0.999, demostrando que son prácticamente el mismo vector. Para hacer este PCA se utilizaron los 585 átomos de Cα de la enzima y si se reemplazan estos datos en la Ec. 13, se encuentra que el RMSD entre las estructuras promedio es de 5.46
, mientras que el único autovalor de la matriz es 4361.59 . El producto escalar entre los dos autovectores se vale 0.999, demostrando que son prácticamente el mismo vector. Para hacer este PCA se utilizaron los 585 átomos de Cα de la enzima y si se reemplazan estos datos en la Ec. 13, se encuentra que el RMSD entre las estructuras promedio es de 5.46 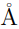 , lo que coincide perfectamente con el obtenido por medios independientes. Obviamente, no hay ninguna necesidad de realizar todo el protocolo de PCA combinado para calcular el RMSD entre dos estructuras promedio, cuando todos los programas de visualización molecular permiten hacer el mismo calculo con solo dos “clicks” en el botón del “mouse”. Respecto al significado del resto de los autovectores de , no es posible hacer predicciones generales ya que los resultados dependen de los pesos relativos de las matrices y , que pueden cambiar de un caso a otro.
, lo que coincide perfectamente con el obtenido por medios independientes. Obviamente, no hay ninguna necesidad de realizar todo el protocolo de PCA combinado para calcular el RMSD entre dos estructuras promedio, cuando todos los programas de visualización molecular permiten hacer el mismo calculo con solo dos “clicks” en el botón del “mouse”. Respecto al significado del resto de los autovectores de , no es posible hacer predicciones generales ya que los resultados dependen de los pesos relativos de las matrices y , que pueden cambiar de un caso a otro.
El caso límite alternativo se encuentra cuando las dos estructuras iniciales pertenecen al mismo pozo de energía libre, de tal modo que ambas trayectorias muestrean el espacio configuracional en forma similar. En ese caso, las estructuras promedio de las dos simulaciones son muy parecidas, por lo que la contribución de la matriz en la Ec. 11 se vuelve despreciable, mientras que las matricesy se asemejan entre sí. Por lo tanto, es de esperar
que los resultados obtenidos al concatenar dos simulaciones independientes sean similares a los de las trayectorias individuales. Si bien esta conclusión es cualitativamente cierta, existen diferencias numéricas que permiten usar la combinación de dos o más simulaciones para mejorar la convergencia de las superficies de energía libre obtenidas por PCA. Discutiremos este tema en la Sección 5.4.
¿Qué podríamos esperar si combinamos más de dos simulaciones? Es evidente que la discusión del párrafo anterior también se aplica a este escenario si las trayectorias utilizadas en la combinación pertenecen al mismo pozo de energía libre. Sin embargo, la discusión es más interesante cuando las trayectorias muestrean regiones distantes del espacio configuracional, de modo que la matriz contribuye en mucho mayor medida a que el promedio de las matrices de covarianza individuales. En la Ref. [37] nosotros presentamos el caso correspondiente a n = 3. Allí mostramos que la matriz 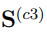 tiene dos autovectores con autovalor distinto de cero. Estos son casi paralelos a los de la matriz
tiene dos autovectores con autovalor distinto de cero. Estos son casi paralelos a los de la matriz 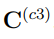 , pero no es posible prever el significado de los demás autovalores y autovectores. Además, los dos primeros autovectores están en el plano que contiene las tres estructuras promedio de las simulaciones individuales, pero no están alineados con ninguna de las tres rectas que unen pares alternativos de estructuras promedio.
, pero no es posible prever el significado de los demás autovalores y autovectores. Además, los dos primeros autovectores están en el plano que contiene las tres estructuras promedio de las simulaciones individuales, pero no están alineados con ninguna de las tres rectas que unen pares alternativos de estructuras promedio.
En el mismo sentido, si combinamos cuatro simulaciones independientes pertenecientes a regiones muy distantes del espacio configuracional, los primeros tres autovectores de 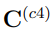 son casi paralelos a los tres autovectores de
son casi paralelos a los tres autovectores de 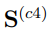 con autovalores distintos de cero. Estos vectores abarcan un espacio tridimensional que contiene las cuatro estructuras promedio de las simulaciones individuales. Entendemos que, a partir de aquí, es fácil prever cómo evolucionarán los resultados si se aumenta el número de simulaciones utilizadas en el PCA combinado, para los dos casos límite considerados hasta ahora. Para concluir, señalamos que, para cualquier caso intermedio donde la matriz y las matrices de covarianza individuales contribuyan de manera similar a , no es posible hacer predicciones generales.
con autovalores distintos de cero. Estos vectores abarcan un espacio tridimensional que contiene las cuatro estructuras promedio de las simulaciones individuales. Entendemos que, a partir de aquí, es fácil prever cómo evolucionarán los resultados si se aumenta el número de simulaciones utilizadas en el PCA combinado, para los dos casos límite considerados hasta ahora. Para concluir, señalamos que, para cualquier caso intermedio donde la matriz y las matrices de covarianza individuales contribuyan de manera similar a , no es posible hacer predicciones generales.
PCA de movimientos ternarios y cuaternarios
Cuando se analizan cambios conformacionales en proteínas multiméricas, suele ser ilustrativo discernir entre los movimientos que modifican la estructura terciaria de la proteína (movimientos intra-cadena) y aquellos que cambian su estructura cuaternaria (movimientos inter-cadena). Sin embargo, si el PCA se realiza sobre la trayectoria directamente obtenida de una simulación MD, los vectores del espacio esencial mezclan las contribuciones de ambos tipos de movimiento, resultando imposible hacer la separación. En el año 2013, de Gert de Groot y sus colaboradores propusieron un método que permite discriminar ambos tipos de movimiento [38], de manera tal que el PCA pueda realizarse sobre cada uno de ellos por separado. A continuación, describimos en qué consiste este procedimiento.
Los movimientos intra-cadena se determinan superponiendo las coordenadas de cada cadena individual correspondientes a los distintos frames sobre una estructura de referencia, que generalmente es la estructura inicial. Por su parte, los movimientos inter-cadena se obtienen haciendo lo opuesto: cada cadena individual de la estructura de referencia se superpone con su cadena correspondiente en los distintos frames. Una representación esquemática del procedimiento se muestra en la Fig. 4. Cuando este procedimiento se aplica a todos los frames colectados de la simulación de MD, se obtienen dos trayectorias ficticias. Una contiene los movimientos intra-cadena y la otra los movimientos inter-cadena. Por construcción, los desplazamientos de los subespacies inter- e intra-cadena son ortogonales entre sí. Además, su suma
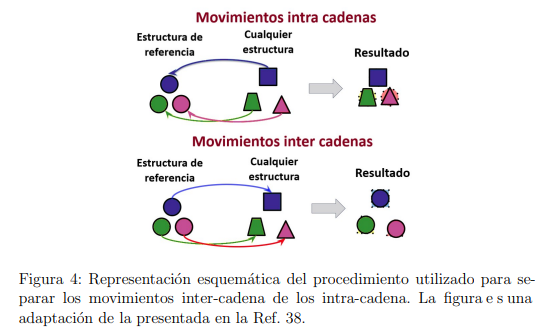
Una vez que se obtuvieron las dos trayectorias ficticias, el procedimiento de PCA se aplica a cada una de ellas por separado. Al hacerlo, se observa inmediatamente que el número de autovectores con autovalores distintos de cero en el PCA inter-cadena es significativamente menor que el del PCA intra-cadena. Esto ocurre porque el número de grados de libertad efectivos de la trayectoria inter-cadena es bastante pequeño. Seis grados de libertad describen las traslaciones y rotaciones globales de cada cadena. Sin embargo, dado que todo el sistema no se traslada ni rota, también hay seis restricciones.
En consecuencia, el número de grados de libertad de los movimientos intercadena es 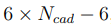 , donde
, donde 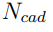 es el número de cadenas de la proteína. De Groot y sus colaboradores emplearon la separación entre modos intercadena e intra-cadena para estudiar el acoplamiento de los movimientos terciarios y cuaternarios en la hemoglobina. Más tarde, en nuestro grupo, lo utilizamos para investigar el mecanismo de apertura del canal P2X4 [39]. Las películas I y II que se presentan como Información Suplementaria, muestran animaciones de los principales autovectores obtenidos al realizar PCA de las trayectorias inter e intra-cadena de P2X4. El análisis reveló que la apertura del poro del canal es causada principalmente por los movimientos inter-cadena. Sin embargo, estos movimientos no pueden lograr la extensión requerida para el pasaje de iones a menos que ocurran deformaciones intra- cadena en la parte superior de la molécula. Por último, debemos notar que, aunque en este apartado hemos discutido la separación inter/intra aplicada a proteínas multiméricas, el procedimiento puede emplearse en muchas otras situaciones. Por ejemplo, podría usarse para analizar los movimientos de diferentes dominios de una proteína dada, o de las diferentes moléculas que componen un complejo.
es el número de cadenas de la proteína. De Groot y sus colaboradores emplearon la separación entre modos intercadena e intra-cadena para estudiar el acoplamiento de los movimientos terciarios y cuaternarios en la hemoglobina. Más tarde, en nuestro grupo, lo utilizamos para investigar el mecanismo de apertura del canal P2X4 [39]. Las películas I y II que se presentan como Información Suplementaria, muestran animaciones de los principales autovectores obtenidos al realizar PCA de las trayectorias inter e intra-cadena de P2X4. El análisis reveló que la apertura del poro del canal es causada principalmente por los movimientos inter-cadena. Sin embargo, estos movimientos no pueden lograr la extensión requerida para el pasaje de iones a menos que ocurran deformaciones intra- cadena en la parte superior de la molécula. Por último, debemos notar que, aunque en este apartado hemos discutido la separación inter/intra aplicada a proteínas multiméricas, el procedimiento puede emplearse en muchas otras situaciones. Por ejemplo, podría usarse para analizar los movimientos de diferentes dominios de una proteína dada, o de las diferentes moléculas que componen un complejo.
Uno de los principales objetivos de PCA es identificar los cambios conformacionales más prominentes de la biomolécula en estudio. Estas conformaciones se modifican cuando varían las distancias entre los átomos de la molécula, pero no cambian cuando la misma se traslada o rota como un cuerpo rígido. Sin embargo, las simulaciones MD que se realizan en coordenadas atómicas cartesianas mezclan estos tipos de movimiento. Por lo tanto, antes de realizar un PCA usando estas coordenadas, es necesario eliminar la influencia de la traslación y rotación global, tal como se indicó en la Sec. 3.1.
El procedimiento se basa en la siguiente idea. Si adosamos un sistema de ejes cartesianos a la molécula, la traslación y la rotación global de la misma pueden ser descritos por los movimientos de dicho sistema de ejes. De ahora en más, nos referiremos al mismo como el sistema de ejes FM por “Fijo en la Molécula”, para distinguirlo de un sistema de ejes que no se traslada ni rota, al que llamaremos FE por “Fijo en el Espacio”. De este modo, la traslación global de la molécula se describe por tres coordenadas cartesianas que señalan el origen del sistema FM desde el origen del sistema FE, mientras que las rotaciones se describen por tres ángulos de Euler que proveen suorientación [34]. La clave del procedimiento reside en definir qué prescripción o receta vamos a utilizar para decir cómo hay que adosar el sistema FM a cada estructura molecular. Por ahora supongamos que hemos resuelto este punto. Entonces, dado un conjunto de estructuras moleculares muestreadas de una trayectoria MD, debemos adosar el sistema FM a cada una de ellas utilizando siempre la misma prescripción. Finalmente, eliminamos la rotación y la traslación global de la molécula alineando el sistema de ejes FM de cada estructura del conjunto con el de una estructura que hayamos elegido como referencia. La trayectoria que se genera al hacer este alineamiento es la que habríamos obtenido si el sistema de ejes FM se hubiera quedado quieto. Así, con buena aproximación, las coordenadas cartesianas resultantes solo dan cuenta de las deformaciones internas de la molécula. Sin embargo, el algoritmo no es exacto y la calidad del resultado depende de los criterios seguidos para adosar el sistema FM a cada estructura molecular muestreada. Veamos entonces, cuáles son los criterios que se suelen aplicar para ello.
El origen del sistema de ejes fijo a la molécula se elige siempre como el centro de masa de la molécula. Con esta elección, la energía cinética de la traslación global se separa de la energía de las rotaciones y de las vibraciones [34]. Sin embargo, no hay una manera única de seleccionar la orientación de los ejes, sino que existen diversas opciones y ninguna de ellas proporciona una separación exacta entre vibración y rotación. Para moléculas pequeñas y/o semirrígidas, el criterio más empleado es la segunda condición de Eckart. Esta alternativa asegura que las pequeñas deformaciones alrededor de la posición de equilibrio de la molécula no contribuyen al momento angular total. Además, esta elección minimiza el acoplamiento entre la energía rotacional y la vibracional [40].
Desafortunadamente, las macromoléculas en solución a temperatura ambiente no son cuerpos semirrígidos, sino que sus superficies de energía libre tienen muchos mínimos locales. Por lo tanto, la segunda condición de Eckart, que asume que hay una única estructura de equilibrio, no puede aplicarse. Una alternativa sería indicar que el sistema FM coincide con los ejes de inercia de la molécula. Sin embargo, en el análisis de simulaciones de MD, el enfoque más comúnmente utilizado es el ajuste de cuadrados mínimos rotacionales presentado por McLachlan [41]. De acuerdo con el mismo, se busca superponer cada una de las estructuras del conjunto con la de referencia, mediante una transformación combinada. Primero, se aplica una traslación que hace coincidir sus centros de masa. Luego, se hace una rotación que minimiza la desviación cuadrática media entre los átomos de ambas estructuras. Se ha demostrado que el ajuste de cuadrados mínimos proporciona la misma alineación que las condiciones de Eckart, si en ambos casos se emplea la misma estructura de referencia [42]. Sin embargo, como se mencionó anteriormente, las macromoléculas biológicas a temperatura ambiente no tienen un único mínimo que pueda seleccionarse inequívocamente como referencia. Para empeorar las cosas, el procedimiento de ajuste de cuadrados mínimos produce resultados ligeramente diferentes si se emplean distintas referencias. Así, la separación aproximada de la rotación funciona bien para moléculas relativamente estructuradas, pero empeora cuanto más flexible sea la molécula en estudio.
5.2 ¿Qué coordenadas me conviene utilizar?
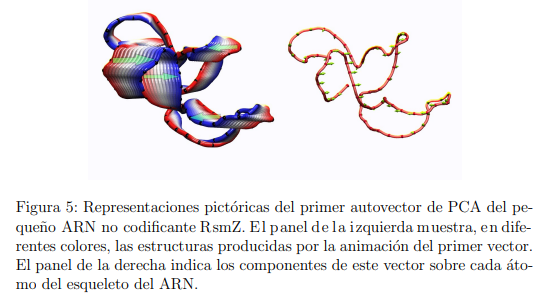
La elección de las coordenadas que conviene utilizar para hacer PCA está estrechamente ligada al propósito del análisis. De lo discutido en la sección anterior se desprende que un PCA realizado en coordenadas cartesianas no es apropiado para ningún trabajo cuantitativo tal como la identificación de las conformaciones estables y metaestables de una biomolécula o la evaluación de sus pesos relativos. Sin embargo, para obtener una representación pictórica de los movimientos relevantes de la molécula se utilizan coordenadas cartesianas. Dicha representación podría ser una imagen que muestre los desplazamientos producidos por un determinado autovector, como el que se muestra en la Fig. 5, o podría ser una película con su animación, como la Película III presentada como Información Suplementaria. Por otra parte, debido al estrecho paralelismo entre PCA y NMA, la mayoría de los programas utilizados para representar o animar modos normales también pueden ser aplicados para ilustrar Componentes Principales. La extensión Normal Mode Wizard del programa VMD es probablemente la más ampliamente empleada para ese propósito [43].
Las dificultades causadas por el uso de coordenadas cartesianas en el PCA de moléculas flexibles han sido estudiadas en detalle en la Ref. 44. En ese estudio determinaron, de manera unívoca, que dichas dificultades surgen de la separación inapropiada entre las deformaciones internas de la molécula y su rotación global, lo que introduce ruido numérico en el análisis. En consecuencia, las superficies de energía libre calculadas a partir de PCA realizadas en coordenadas cartesianas tienen características artificiales que impiden la identificación de los estados estables y metaestables del sistema. Estos problemas se han encontrado incluso en el PCA de una proteína tan rígida como BPTI, cuando se utilizó una trayectoria muy larga [44]. Para superar estos inconvenientes se han ideado esquemas de superposición sofisticados [45] que mejoran el análisis en algunos casos, pero no en todos [44]. Alternativamente, todas las dificultades se pueden evitar si se lleva a cabo el PCA en coordenadas internas, tales como distancias y/o ángulos diedros. Como discutiremos a continuación, cada una de estas opciones se adapta a un tipo particular de problema y tiene sus propias limitaciones y deficiencias.
Para un conjunto de N puntos, hay 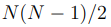 distancias entre pares, mientras que el número de grados de libertad internos, o sea los grados de libertad que describen las deformaciones de la nube de puntos, es
distancias entre pares, mientras que el número de grados de libertad internos, o sea los grados de libertad que describen las deformaciones de la nube de puntos, es 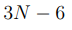 . Por lo tanto, para
. Por lo tanto, para 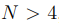 , hay más distancias que coordenadas internas independientes. Además, el número de distancias crece muy rápidamente con N y pronto se convierte en un número enorme. 9 Esto hace que la diagonalización de la matriz de covarianza, requerida para hacer PCA, se vuelva computacionalmente muy costosa o incluso sea imposible de realizar. Por lo expuesto, antes de hacer un PCA basado en distancias, uno debe elegir cuidadosamente cuáles se van a incluir en el análisis. Las opciones típicas son utilizar las distancias entre carbonos alfa o las distancias entre los átomos más próximos de cada par de residuos. Sin embargo, incluso este conjunto suele ser demasiado grande para una implementación numérica eficiente [46], por lo que se suelen agregar otros criterios para reducirlo aún más. Para tal fin, el criterio más utilizado es emplear solo las distancias entre residuos que están en contacto en la estructura nativa [47]. Además, las distancias entre residuos que están separados por menos de cuatro residuos en la secuencia proteica también se descartan.
, hay más distancias que coordenadas internas independientes. Además, el número de distancias crece muy rápidamente con N y pronto se convierte en un número enorme. 9 Esto hace que la diagonalización de la matriz de covarianza, requerida para hacer PCA, se vuelva computacionalmente muy costosa o incluso sea imposible de realizar. Por lo expuesto, antes de hacer un PCA basado en distancias, uno debe elegir cuidadosamente cuáles se van a incluir en el análisis. Las opciones típicas son utilizar las distancias entre carbonos alfa o las distancias entre los átomos más próximos de cada par de residuos. Sin embargo, incluso este conjunto suele ser demasiado grande para una implementación numérica eficiente [46], por lo que se suelen agregar otros criterios para reducirlo aún más. Para tal fin, el criterio más utilizado es emplear solo las distancias entre residuos que están en contacto en la estructura nativa [47]. Además, las distancias entre residuos que están separados por menos de cuatro residuos en la secuencia proteica también se descartan.
Por su parte, para el PCA basado en diedros, se pueden utilizar los ángulos del esqueleto de la molécula o también los de las cadenas laterales. La primera opción es, por mucho, la más utilizada, porque permite revelar los cambios conformacionales más significativos de la molécula. En cambio, si se hace un PCA de diedros de las cadenas laterales, lo que se observa son variaciones de los contactos entre residuos, pero el PCA basado en distancias de contacto es claramente una herramienta más adecuada para este fin [47].
Antes de realizar un PCA basado en ángulos diedros, uno debe tener en cuenta los problemas que pueden originarse de la periodicidad de los mismos. Debido a esta periodicidad, por ejemplo, puntos muy cercanos como 179° y -179° están separados artificialmente cuando el rango angular utilizado es (-180°:180°]. Esta separación artificial persiste, aunque se cambie el rango utilizado, porque dicho cambio solo modifica la región en la que se encontraran las dificultades. Por ejemplo, al usar [0°:360°), no hay problemas alrededor de 180° porque los mismos se han desplazado a alrededor de 0°/360°. A veces ocurre que los ángulos utilizados no muestrean todo el intervalo de 360°.10 En tales circunstancias, uno puede elegir el intervalo angular apropiado para cada caso, de manera tal que las discontinuidades aparezcan en las regiones que no son muestreadas por el sistema. En esta idea se basa el método llamado de dPCA+, desarrollado por Gerard Stock y colaboradores [48]. Una ilustración de cómo se realiza el cambio en el intervalo angular se proporciona en la Fig. 3 de la Ref. 48. Sin embargo, la estrategia de dPCA+ es engorrosa cuando uno debe lidiar con muchos ángulos diferentes, ya que requiere determinar el rango apropiado para cada uno de ellos. Además, para algunas moléculas y algunos ángulos en particular, puede suceder que todo el intervalo angular sea efectivamente visitado. Esto hace que dPCA+ sea inútil.
El procedimiento más ampliamente empleado para realizar PCA de ángulos diedros se llama dPCA y fue desarrollado por Stock varios años antes que dPCA+ [49]. El algoritmo propone duplicar el número de variables mediante la transformación,

donde n toma valores desde 1 hasta el número total de ángulos considerados. Esta alternativa tiene la clara desventaja de aumentar artificialmente el tamaño de las matrices con las que uno tiene que lidiar, pero también tiene diversas ventajas que explican su “popularidad”. Principalmente, con dPCA se evitan las discontinuidades que surgen de la periodicidad de los ángulos y se elimina el problema de los promedios mal definidos. Este último inconveniente afecta gravemente el cálculo de la matriz de covarianza. Para entender su origen, consideraremos dos ángulos, uno que oscila alrededor de 0°, entre -10° y 10°; y el otro que oscila alrededor de 180°, entre -170° y 170°. Dado que el promedio se sitúa en 0° en ambos casos, las desviaciones respecto del promedio, que se utilizan para calcular la matriz de covarianza, resultan ser mucho mayores en el segundo que en el primer caso. Sin embargo, esta diferencia no refleja la verdadera situación física, ya que en los dos casos los desplazamientos son de 10° respecto al promedio. Afortunadamente, el método dPCA elimina naturalmente todos estos inconvenientes. Además, provee un mapeo biunívoco de la distribución angular original. Esto significa que no crea ni suprime mínimos en las superficies de energía libre de la molécula. Finalmente, el procedimiento puede ser fácilmente sistematizado y constituye una de las opciones disponibles para realizar PCA en el marco de la gran mayoría de los programas que analizan simulaciones MD.
La experiencia con el análisis de diferentes sistemas indica que dPCA es el método de elección cuando se pretenden identificar los estados estables y metaestables de moléculas que experimentan grandes cambios conformacionales. Esto se aplica, por ejemplo, a estudios de plegamiento de proteínas, así como a cambios conformacionales de ARNs [50]. Por otro lado, si uno tiene como objetivo caracterizar los estados de proteínas estructuradas que llevan a cabo sus movimientos funcionales, el PCA basado en distancias resulta ser el más conveniente [47]. En cualquier caso, siempre se deben probar las diferentes alternativas, comparar sus resultados e intentar evaluar la calidad de sus predicciones mediante procedimientos independientes. Para concluir, señalamos que la Ref. 51 ofrece una discusión muy clara y detallada de las ventajas y desventajas de las diferentes coordenadas utilizadas para llevar a cabo PCA de macromoléculas. Recomendamos encarecidamente la lectura de ese artículo a aquellos que buscan profundizar en los temas presentados en esta sección.
5.3 ¿Cómo se determina el tamaño del espacio esencial?
Hasta este punto, hemos estado hablando acerca del ES, una pequeña subespecie vectorial que contiene las direcciones de las fluctuaciones más importantes de la molécula bajo estudio. Sin embargo, no hemos indicado aun ningún criterio que nos permita decidir cuántos autovectores deben incluirse en dicho espacio. De hecho, se pueden aplicar diversos criterios y la elección del mismo depende del propósito que se persigue al hacer el PCA.
Como se mencionó anteriormente, los autovalores obtenidos por PCA son las fluctuaciones cuadráticas medias de los Componentes Principales. Asimismo, al realizar el PCA en coordenadas cartesianas, la suma de todas estas fluctuaciones equivale a la suma de las fluctuaciones cuadráticas de los átomos incluidos en el análisis. Así, uno de los criterios más simples para decidir el tamaño del ES consiste en incluir los primeros n autovectores de PCA, de modo que estos den cuenta de una fracción dada de las fluctuaciones cuadráticas totales. Por lo tanto, si f es la fracción de las fluctuaciones que se desea tener en cuenta, y el PCA se realizó utilizando las coordenadas cartesianas de átomos, el número n de autovectores en el ES se elige de manera que,

Así, por ejemplo, para los datos mostrados en la Fig. 1, elegir f = 0,9 conduce a un ES con 12 autovectores mientras que establecer f = 0,75 incluye solo 5 autovectores.
9Esto se debe a su dependencia cuadrática con N. Si se duplica el número de puntos, prácticamente se cuadruplica el número de distancias.
10Esto ocurre muchas veces con los ángulos diedros del esqueleto de las proteínas plegadas.
Una idea menos arbitraria para determinar el espacio esencial tiene como objetivo detectar los Componentes Principales que no describen movimientos triviales, entendiendo por triviales a aquellos movimientos que son prácticamente armónicos. Para ello, las estructuras muestreadas de las simulaciones MD se proyectan sobre los primeros autovectores de la matriz de covarianza (Ec.6) a fin de obtener el conjunto, 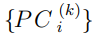 , que contiene los valores muestreados por el Componente Principal i a lo largo de dicha trayectoria. Luego, con los datos de cada conjunto, se calcula y grafica un histograma o la función densidad de probabilidad. Típicamente, las distribuciones de los primeros PCs tienen formas no-gaussianas mientras que, a medida que crece el índice del Componente Principal, sus formas se asemejan más y más a la de una gaussiana. De acuerdo con esto, el ES se forma con todos los Componentes Principales cuyas distribuciones difieren notoriamente de una gaussiana.
, que contiene los valores muestreados por el Componente Principal i a lo largo de dicha trayectoria. Luego, con los datos de cada conjunto, se calcula y grafica un histograma o la función densidad de probabilidad. Típicamente, las distribuciones de los primeros PCs tienen formas no-gaussianas mientras que, a medida que crece el índice del Componente Principal, sus formas se asemejan más y más a la de una gaussiana. De acuerdo con esto, el ES se forma con todos los Componentes Principales cuyas distribuciones difieren notoriamente de una gaussiana.
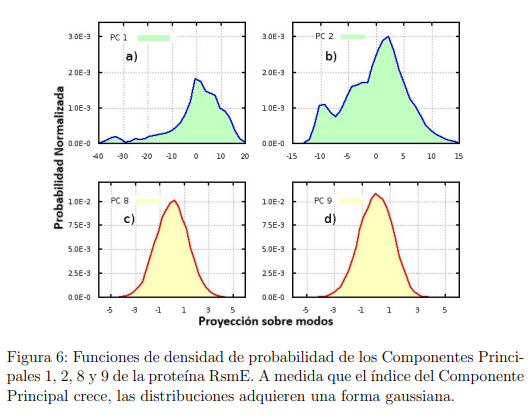
En la Fig.6 se muestran las distribuciones obtenidas para los Componentes Principales 1, 2, 8 y 9 de la proteína RsmE. Es importante destacar que este criterio basado en las distribuciones, debe utilizarse solo después de haber asegurado la convergencia del cálculo de PCA (que se discute en la sección próxima). Ocurre que muchas veces existen correlaciones fortuitas entre las coordenadas atómicas muestreadas en simulaciones MD. Al decir fortuitas nos referimos a correlaciones que no son causadas por la superficie de energía potencial, sino que ocurren al azar. En un artículo recientemente aceptado para su publicación, nosotros discutimos el efecto de estas correlaciones en el análisis de PCA. Y uno de sus efectos más dañinos es la aparición de distribuciones aparentemente no-gaussianas que se vuelven gaussianas al converger el cálculo [13].
Finalmente, otro de los criterios ampliamente utilizados para determinar el número de autovectores a incluir en el ES es el del scree plot [52]. Un scree plot es un gráfico de los autovalores de PCA en función de su ´índice, como se muestra en la curva roja de la Fig.1. Literalmente, el término scree se refiere a los escombros que se acumulan al pie de una montaña o acantilado, y en este caso hace referencia a la forma que típicamente adopta un gráfico de autovalores de PCA versus índice. Todos ellos disminuyen rápidamente al principio, pero luego se estabilizan y continúan disminuyendo mucho más lentamente, demostrando que unos pocos autovectores tienen desplazamientos importantes. Con base en esta observación, el procedimiento consiste en buscar el punto donde el scree plot tiene un cambio nítido de pendiente. Todos los puntos hasta ese punto se incluyen en el ES mientras que los que tienen índices más altos se descartan. Con este criterio, el ES del grafico de la Fig.1 solo tendría 2 vectores.
5.4 ¿Cómo sé si mis resultados están convergidos?
La preocupación más común al analizar los resultados de simulaciones de dinámica molecular es saber si están convergidos o no. Esta preocupación ha inquietado a los realizadores de estas simulaciones desde las primeras aplicaciones de la metodología hasta el presente, a pesar de que los tiempos de simulación alcanzables se multiplicaron por casi 106. Una discusión exhaustiva de este problema excede el alcance de este artículo, pero remitimos a los lectores interesados a las referencias Ref. 53, 54 y 55. Allí podrán encontrar una discusión profunda del tema, mientras que en este texto nos limitaremos a analizar las principales tendencias observadas, para luego enfocarnos en las propiedades de convergencia de PCA.
Cuanto más grande es una macromolécula, mayor es el tamaño de su espacio configuracional. Por lo tanto, en principio, las simulaciones de dinámica molecular de moléculas grandes son más difíciles de converger que las de moléculas pequeñas. Sin embargo, la flexibilidad de la molécula también juega un papel. Las macromoléculas biológicas solo visitan una fracción del espacio configuracional disponible. Cuanto más fuertes sean las interacciones entre sus componentes, más correlacionados serán sus movimientos y menor será la fracción del espacio configuracional accesible, lo que mitiga sustancialmente los requisitos de muestreo. Por esta razón, las simulaciones MD de proteínas plegadas convergen mucho más fácilmente que aquellas que buscan simular los procesos de plegamiento. Por lo mismo, es difícil muestrear adecuadamente el espacio configuracional de proteínas intrínsecamente desordenadas o de fragmentos de ARN. Por último, debemos destacar que la cantidad de muestreo requerida depende del parámetro que se quiere calcular. Algunas cantidades, como la energía interna o la entalpía, convergen más rápido que la entropía y la energía libre. Esto ocurre porque las simulaciones MD visitan frecuentemente las regiones de baja energía potencial que son las que más contribuyen a la energía interna. La entropía, en cambio, requiere evaluar todo el volumen del espacio configuracional accesible, con la particularidad de que las regiones poco visitadas y las muy visitadas hacen contribuciones del mismo orden de magnitud. Como consecuencia de esta característica, los cálculos de entropía convergen de forma extremadamente lenta. Por ende, lo mismo ocurre con la energía libre.
Para evaluar la convergencia de los resultados de PCA de macromoléculas biológicas se desarrollaron diversos criterios. El más estricto consiste en calcular el producto escalar entre los vectores del espacio esencial obtenidos de trayectorias independientes, calculadas en las mismas condiciones. Si rotulamos a estas trayectorias equivalentes como (a) y (b), la evaluación consiste en calcular 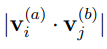 . El resultado debería ser 1 para
. El resultado debería ser 1 para 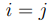 y 0 en caso contrario. Una práctica común es presentar estos productos en forma de matriz, indicando el valor absoluto del producto escalar calculado como el radio de un círculo centrado en cada punto (i, j). Estas son las llamadas “matrices de producto interno” que se muestran, por ejemplo, en la Fig. 2 de la Ref. 56. En nuestro grupo analizamos en detalle las propiedades de convergencia de los primeros autovectores, dado que estos son los más importantes. Para ello, calculamos el producto
y 0 en caso contrario. Una práctica común es presentar estos productos en forma de matriz, indicando el valor absoluto del producto escalar calculado como el radio de un círculo centrado en cada punto (i, j). Estas son las llamadas “matrices de producto interno” que se muestran, por ejemplo, en la Fig. 2 de la Ref. 56. En nuestro grupo analizamos en detalle las propiedades de convergencia de los primeros autovectores, dado que estos son los más importantes. Para ello, calculamos el producto 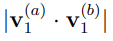 para todos los pares del primer autovectores que pudimos formar a partir de 180 de trayectorias equivalentes de la proteína BPTI [56]. Luego estimamos la densidad de probabilidad de estos productos escalares. La función resultante se comparó con la correspondiente al producto escalar de vectores de igual dimensionalidad pero de direcciones aleatorias [57]. El procedimiento reveló que los productos obtenidos de simulaciones de MD suelen ser significativamente mayores que los de vectores aleatorios. No obstante, se pueden obtener productos escalares muy bajos, lo que indica que los Componentes Principales de las trayectorias equivalentes son prácticamente ortogonales entre sí. Este comportamiento constituye una muestra clara de que el análisis no está convergido y por lo tanto, de que los vectores obtenidos no guardan relación con la superficie de energía libre que gobierna los movimientos moleculares.
para todos los pares del primer autovectores que pudimos formar a partir de 180 de trayectorias equivalentes de la proteína BPTI [56]. Luego estimamos la densidad de probabilidad de estos productos escalares. La función resultante se comparó con la correspondiente al producto escalar de vectores de igual dimensionalidad pero de direcciones aleatorias [57]. El procedimiento reveló que los productos obtenidos de simulaciones de MD suelen ser significativamente mayores que los de vectores aleatorios. No obstante, se pueden obtener productos escalares muy bajos, lo que indica que los Componentes Principales de las trayectorias equivalentes son prácticamente ortogonales entre sí. Este comportamiento constituye una muestra clara de que el análisis no está convergido y por lo tanto, de que los vectores obtenidos no guardan relación con la superficie de energía libre que gobierna los movimientos moleculares.
En muchas situaciones, las evaluaciones de convergencia discutidas anteriormente son demasiado estrictas. Usualmente no es necesario que dos simulaciones independientes proporcionen exactamente los mismos autovectores, sino que los vectores de ambos ESs expandan la misma subespecio. Si esta condición se cumple, los dos subespecios son capaces de describir las mismas deformaciones internas. Un parámetro que mide la similitud entre dos subespacios alternativos se llama RMSIP, que es una sigla que viene del inglés Root Mean Squared Inner Product y que se calcula como,
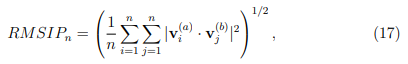
donde n es el número de vectores del espacio esencial considerado. 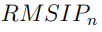 es igual a 1 cuando los dos espacios esenciales abarcan el mismo subespacio, mientras que vale 0 si los mismos son ortogonales entre sí. Las primeras evaluaciones de convergencia de PCA de proteínas consistieron en dividir la trayectoria en dos mitades y calcular entre ambas partes, tomando porciones de longitud temporal creciente. Típicamente, estas curvas aumentan rápidamente al principio, pero luego se nivelan alcanzando un valor de plateau menor que 1.0. Estas formas parecen indicar que la consistencia de los resultados no se puede mejorar más allá del valor alcanzado en el plateau (ver por ejemplo Fig. 1 de la Ref. [57]). Como veremos a continuación, afortunadamente este no es el caso.
es igual a 1 cuando los dos espacios esenciales abarcan el mismo subespacio, mientras que vale 0 si los mismos son ortogonales entre sí. Las primeras evaluaciones de convergencia de PCA de proteínas consistieron en dividir la trayectoria en dos mitades y calcular entre ambas partes, tomando porciones de longitud temporal creciente. Típicamente, estas curvas aumentan rápidamente al principio, pero luego se nivelan alcanzando un valor de plateau menor que 1.0. Estas formas parecen indicar que la consistencia de los resultados no se puede mejorar más allá del valor alcanzado en el plateau (ver por ejemplo Fig. 1 de la Ref. [57]). Como veremos a continuación, afortunadamente este no es el caso.
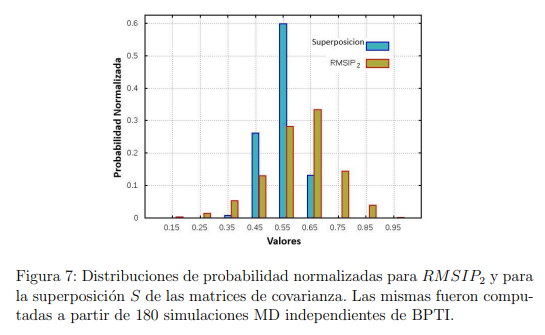
Finalmente, otro parámetro utilizado para evaluar la convergencia de los PCA de simulaciones MD es la llamada “superposición de covarianza”, S, propuesta por Hess en 2002 [58]. En realidad, S no es una medida de la convergencia del ES, sino que evalúa la similitud de los espacios muestreados por un par de trayectorias, mediante una comparación de sus matrices de covarianza. Esta superposición se define como,
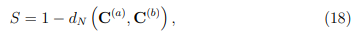
donde 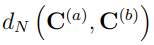 es la distancia normalizada entre las matrices de covarianza
es la distancia normalizada entre las matrices de covarianza 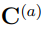 y
y 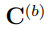 , la cual se calcula como
, la cual se calcula como
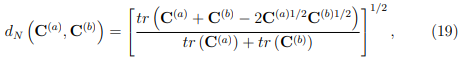
donde 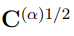 , la raíz cuadrada de la matriz de correlación
, la raíz cuadrada de la matriz de correlación 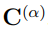 . Si dos simulaciones diferentes proporcionan el mismo muestreo, sus matrices de covarianza son iguales, la distancia entre ellas es cero y su superposición vale
. Si dos simulaciones diferentes proporcionan el mismo muestreo, sus matrices de covarianza son iguales, la distancia entre ellas es cero y su superposición vale
1. Por el contrario, si las simulaciones han muestreado subespacios que son ortogonales entre sí, la distancia normalizada vale 1 y S vale cero.
La Fig. 7 muestra las funciones de distribución de probabilidad para 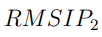 y S calculadas a partir de trayectorias de 50 ns de la proteína BPTI. Para obtener este gráfico, calculamos 80 trayectorias de BPTI utilizando la misma estructura inicial y las mismas condiciones (T , P y otros parámetros requeridos para hacer las simulaciones). La diferencia entre ellas reside solo en las velocidades iniciales de los átomos, las que fueron elegidas al azar desde una distribución de Maxwell-Boltzmann a la temperatura correspondiente. Luego, para cada una de estas trayectorias, realizamos PCA y evaluamos y S para los 3160 pares de trayectorias que es posible comparar. La figura muestra que, en algunos casos, se pueden obtener fortuitamente muy buenos valores para estos parámetros. Sin embargo, también son posibles resultados muy pobres. Cabe mencionar que si se emplean trayectorias más cortas, los resultados son peores. Un análisis similar realizado con la enzima Lisozima arrojó las mismas conclusiones [56].
y S calculadas a partir de trayectorias de 50 ns de la proteína BPTI. Para obtener este gráfico, calculamos 80 trayectorias de BPTI utilizando la misma estructura inicial y las mismas condiciones (T , P y otros parámetros requeridos para hacer las simulaciones). La diferencia entre ellas reside solo en las velocidades iniciales de los átomos, las que fueron elegidas al azar desde una distribución de Maxwell-Boltzmann a la temperatura correspondiente. Luego, para cada una de estas trayectorias, realizamos PCA y evaluamos y S para los 3160 pares de trayectorias que es posible comparar. La figura muestra que, en algunos casos, se pueden obtener fortuitamente muy buenos valores para estos parámetros. Sin embargo, también son posibles resultados muy pobres. Cabe mencionar que si se emplean trayectorias más cortas, los resultados son peores. Un análisis similar realizado con la enzima Lisozima arrojó las mismas conclusiones [56].
En la Ref. [56], nosotros demostramos que la consistencia del PCA calculado a partir de simulaciones de MD puede ser mejorado mediante la combinación de trayectorias independientes, pero equivalentes, de acuerdo con el procedimiento descrito en la Sección 4.1. En principio, uno podría creer que esto ocurre porque la matriz de covarianza obtenida combinando n trayectorias siempre puede escribirse como el promedio de n matrices de covarianza independientes. De acuerdo con esto, al aumentar n, la incertidumbre estadística de los elementos de la matriz de covarianza se reduce por factor 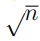 . Sin embargo hay una razón más profunda que explica la efectividad del procedimiento. Ocurre que, en las simulaciones individuales, se crean correlaciones fortuitas entre las coordenadas atómicas. Decimos que estas correlaciones son fortuitas porque no están originadas en la forma de la superficie de energía potencial, sino que son consecuencia del tamaño enorme que tiene el espacio configuracional de las macromoléculas. Hay tantas coordenadas que, por casualidad, algunas de ellas varían en forma concertada. Como dijimos anteriormente, el método de PCA genera nuevas coordenadas buscando que las mismas contengan la mayor fracción posible de las correlaciones observadas en la simulación. Sin embargo, no puede distinguir si las mismas son fortuitas o están determinadas por el potencial. No obstante, cuando se determinan muchas trayectorias equivalentes, ocurre que las correlaciones fortuitas de unas y otras difieren. Y, por lo tanto, cuando se genera una trayectoria ficticia mediante la concatenación de simulaciones independientes, las correlaciones fortuitas desaparecen y solo quedan las originadas en el potencial. En nuestra experiencia, trabajando con sistemas alternativos, este procedimiento requiere un número bastante alto de trayectorias
. Sin embargo hay una razón más profunda que explica la efectividad del procedimiento. Ocurre que, en las simulaciones individuales, se crean correlaciones fortuitas entre las coordenadas atómicas. Decimos que estas correlaciones son fortuitas porque no están originadas en la forma de la superficie de energía potencial, sino que son consecuencia del tamaño enorme que tiene el espacio configuracional de las macromoléculas. Hay tantas coordenadas que, por casualidad, algunas de ellas varían en forma concertada. Como dijimos anteriormente, el método de PCA genera nuevas coordenadas buscando que las mismas contengan la mayor fracción posible de las correlaciones observadas en la simulación. Sin embargo, no puede distinguir si las mismas son fortuitas o están determinadas por el potencial. No obstante, cuando se determinan muchas trayectorias equivalentes, ocurre que las correlaciones fortuitas de unas y otras difieren. Y, por lo tanto, cuando se genera una trayectoria ficticia mediante la concatenación de simulaciones independientes, las correlaciones fortuitas desaparecen y solo quedan las originadas en el potencial. En nuestra experiencia, trabajando con sistemas alternativos, este procedimiento requiere un número bastante alto de trayectorias  para alcanzar una consistencia casi perfecta en los resultados. Sin embargo, solo un puñado de ellas, es suficiente para asegurar que los resultados no son muy pobres (casi completamente aleatorios). Para concluir, debemos señalar que al hablar de “consistencia del ES” no nos referimos a la convergencia de los autovectores con respecto al tiempo, sino a obtener el mismo resultado cuando se realiza el mismo experimento computacional. La convergencia con respecto al tiempo requiere que todas las regiones del espacio configuracional se hayan muestreado en la proporción correcta. Como se discute en la Ref. 53, este objetivo es mucho más difícil de lograr.
para alcanzar una consistencia casi perfecta en los resultados. Sin embargo, solo un puñado de ellas, es suficiente para asegurar que los resultados no son muy pobres (casi completamente aleatorios). Para concluir, debemos señalar que al hablar de “consistencia del ES” no nos referimos a la convergencia de los autovectores con respecto al tiempo, sino a obtener el mismo resultado cuando se realiza el mismo experimento computacional. La convergencia con respecto al tiempo requiere que todas las regiones del espacio configuracional se hayan muestreado en la proporción correcta. Como se discute en la Ref. 53, este objetivo es mucho más difícil de lograr.
En este artículo hemos discutido cuál es la motivación para realizar un PCA con los datos recopilados de una simulación de MD. Además, hemos indicado cómo interpretar los resultados. Se han presentado las principales deficiencias del MD-PCA y se han proporcionado varios consejos para mitigar el efecto de las mismas. Para finalizar, presentamos a continuación una lista de consejos que recomendamos seguir para lograr un uso provechoso de esta técnica.
* El primer paso para hacer un MD-PCA es seleccionar qué partes de la molécula se van a incluir en el análisis. Todas aquellas regiones que realizan grandes movimientos de carácter arbitrario deben descartarse. Los N- y C- terminales de las proteínas son ejemplos típicos de esto.
* El PCA de coordenadas cartesianas debe usarse principalmente con fines ilustrativos, tales como observar visualmente las deformaciones producidas por los principales autovectores.
* Al estudiar moléculas que experimentan cambios conformacionales significativos, tales como ARN, proteínas intrínsecamente desordenadas, o para simular procesos de plegamiento de proteínas, cualquiera de las variantes de PCA de ángulos diedros del esqueleto molecular representa la mejor opción.
* Las distancias de contacto o los ángulos diedros de las cadenas laterales son coordenadas apropiadas para caracterizar los cambios conformacionales de las proteínas plegadas.
* Al estudiar proteínas multímeras, a menudo es conveniente separar los movimientos inter-cadena de los intra-cadena, para luego realizar un PCA en ambas trayectorias separadas.
* La reproducibilidad del ES determinado a partir de MD-PCA puede mejorarse sustancialmente realizando PCA sobre una trayectoria ficticia que se forma concatenando trayectorias independientes, pero equivalentes, del sistema en cuestión. Aunque se requieren muchas simulaciones para obtener resultados consistentes, combinar solo un puñado de ellas evita que el espacio esencial esté mal definido. Por lo tanto recomendamos fuertemente hacer, siempre, PCA de trayectorias concatenadas.
Hervé Abdi and Lynne J Williams. Principal component analysis. Wiley interdisciplinary reviews: computational statistics, 2(4):433–459, 2010.
Svante Wold, Kim Esbensen, and Paul Geladi. Principal component analysis. Chemometrics and Intelligent Laboratory Systems, 2(1):37–52, 1987. Proceedings of the Multivariate Statistical Workshop for Geolo- gists and Geochemists.
Karl Pearson. Liii. on lines and planes of closest fit to systems of points in space. The London, Edinburgh, and Dublin philosophical magazine and journal of science, 2(11):559–572, 1901.
Harold Hotelling. Analysis of a complex of statistical variables into principal components. Journal of educational psychology, 24(6):417, 1933.
M Alex O Vasilescu and Demetri Terzopoulos. Multilinear image analysis for facial recognition. In Object recognition supported by user interaction for service robots, volume 2, pages 511–514. IEEE, 2002.
Y Vijaya Lata, Chandra Kiran Bharadwaj Tungathurthi, H Ram Mohan Rao, A Govardhan, and LP Reddy. Facial recognition using eigenfaces by pca. International Journal of Recent Trends in Engineering, 1(1):587, 2009.
David Reich, Alkes L Price, and Nick Patterson. Principal component analysis of genetic data. Nature genetics, 40(5):491–492, 2008.
Qian Du and James E Fowler. Hyperspectral image compression using jpeg2000 and principal component analysis. IEEE Geoscience and Re- mote sensing letters, 4(2):201–205, 2007.
M. Pechenizkiy, A. Tsymbal, and S. Puuronen. Pca-based feature transformation for classification: issues in medical diagnostics. In Proceedings. 17th IEEE Symposium on Computer-Based Medical Systems, pages 535– 540, 2004.
MP Robertson, N Caithness, and MH Villet. A pca-based modelling technique for predicting environmental suitability for organisms from presence records. Diversity and distributions, 7(1-2):15–27, 2001.
Jiaoyan Huang, Hyun-Deok Choi, Philip K Hopke, and Thomas M Holsen. Ambient mercury sources in rochester, ny: results from principle components analysis (pca) of mercury monitoring network data. Environmental science & technology, 44(22):8441–8445, 2010.
Juliana Palma and Gustavo Pierdominici-Sottile. On the uses of pca to characterise molecular dynamics simulations of biological macromolecules: Basics and tips for an effective use. ChemPhysChem, 24(2), 2023.
Juliana Palma and Gustavo Pierdominici-Sottile. Fortuitous correlations in molecular dynamics simulations: Their harmful influence on the probability distributions of the main principal components. accepted for publication in ACS Omega, April 2024.
Angel E. García. Large-amplitude nonlinear motions in proteins. Phys. Rev. Lett., 68:2696–2699, Apr 1992.
Andrea Amadei, Antonius BM Linssen, and Herman JC Berendsen. Essential dynamics of proteins. Proteins: Structure, Function, and Bioinformatics, 17(4):412–425, 1993.
Jochen S. Hub and Bert L. de Groot. Detection of functional modes in protein dynamics. PLOS Computational Biology, 5(8):1–13, 08 2009.
German P Barletta and Sebastian Fernandez-Alberti. Protein fluctuations and cavity changes relationship. Journal of Chemical Theory and Computation, 14(2):998–1008, 2018.
Gia G Maisuradze, Adam Liwo, and Harold A Scheraga. Principal component analysis for protein folding dynamics. Journal of molecular bio- logy, 385(1):312–329, 2009.
Elena Papaleo, Paolo Mereghetti, Piercarlo Fantucci, Rita Grandori, and Luca De Gioia. Free-energy landscape, principal component analysis, and structural clustering to identify representative conformations from molecular dynamics simulations: The myoglobin case. Journal of Molecular Graphics and Modelling, 27(8):889–899, 2009.
Jürgen Schlitter. Estimation of absolute and relative entropies of macromolecules using the covariance matrix. Chemical Physics Letters, 215(6):617–621, 1993.
Alfredo Di Nola, Herman JC Berendsen, and Olle Edholm. Free energy determination of polypeptide conformations generated by molecular dynamics. Macromolecules, 17(10):2044–2050, 1984.
Ioan Andricioaei and Martin Karplus. On the calculation of entropy from covariance matrices of the atomic fluctuations. The Journal of Chemical Physics, 115(14):6289–6292, 2001.
Phuong H. Nguyen and Philippe Derreumaux. Configurational entropy: an improvement of the quasiharmonic approximation using configurational temperature. Phys. Chem. Chem. Phys., 14:877–886, 2012.
Kyle W Harpole and Kim A Sharp. Calculation of configurational entropy with a boltzmann–quasiharmonic model: the origin of high- affinity protein–ligand binding. The Journal of Physical Chemistry B, 115(30):9461–9472, 2011.
Riccardo Baron, Philippe H Hünenberger, and J Andrew McCammon. Absolute single-molecule entropies from quasi-harmonic analysis of microsecond molecular dynamics: correction terms and convergence properties. Journal of chemical theory and computation, 5(12):3150–3160, 2009.
Jorge Numata, Michael Wan, and Ernst-Walter Knapp. Conformational entropy of biomolecules: beyond the quasi-harmonic approximation. Genome Informatics, 18:192–205, 2007.
David CC and Jacobs DJ. Principal component analysis: a method for determining the essential dynamics of proteins. Methods Mol Biol., 1084:193–2263, 2014. PMID: 240619231.
Sarah A. Mueller Stein, Anne E. Loccisano, Steven M. Firestine, and Jeffrey D. Evanseck. Chapter 13 principal components analysis: A review of its application on molecular dynamics data. In David C. Spellmeyer, editor, Annual Reports in Computational Chemistry, volume 2, pages 233–261. Elsevier, 2006.
Akio Kitao and Nobuhiro Go. Investigating protein dynamics in collective coordinate space. Current Opinion in Structural Biology, 9(2):164– 169, 1999.
Isabella Daidone and Andrea Amadei. Essential dynamics: foundation and applications. WIREs Computational Molecular Science, 2(5):762– 770, 2012.
Herman JC Berendsen and Steven Hayward. Collective protein dynamics in relation to function. Current Opinion in Structural Biology, 10(2):165–169, 2000.
Miguel L. Teodoro, George N. Phillips, and Lydia E. Kavraki. Unders- tanding protein flexibility through dimensionality reduction. Journal of Computational Biology, 10(3-4):617–634, 2003. PMID: 12935348.
Steven Hayward and Bert L. de Groot. Normal Modes and Essential Dynamics, pages 89–106. Humana Press, Totowa, NJ, 2008.
H. Goldstein, C.P. Poole, and J.L. Safko. Classical Mechanics. Addison Wesley, 2002.
Ian T. Jolliffe and Jorge Cadima. Principal component analysis: a review and recent developments. Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences, 374(2065):20150202, 2016.
I.T. Jolliffe. Principal Component Analysis. Springer Verlag, 1986.
Gustavo Pierdominici-Sottile and Juliana Palma. New insights into the meaning and usefulness of principal component analysis of concatenated trajectories. Journal of Computational Chemistry, 36(7):424–432, 2015.
Martin D Vesper and Bert L De Groot. Collective dynamics underlying allosteric transitions in hemoglobin. PLoS computational biology, 9(9):e1003232, 2013.
Gustavo Pierdominici-Sottile, Luciano Moffatt, and Juliana Palma. The dynamic behavior of the p2x4 ion channel in the closed conformation. Biophysical journal, 111(12):2642–2650, 2016.
Carl Eckart. Some studies concerning rotating axes and polyatomic molecules. Phys. Rev., 47:552–558, Apr 1935.
Andrew D McLachlan. A mathematical procedure for superimpo- sing atomic coordinates of proteins. Acta Crystallographica Section A: Crystal Physics, Diffraction, Theoretical and General Crystallography, 28(6):656–657, 1972.
Konstantin N Kudin and Anatoly Y Dymarsky. Eckart axis conditions and the minimization of the root-mean-square deviation: Two closely related problems. The Journal of chemical physics, 122(22):224105, 2005.
William Humphrey, Andrew Dalke, and Klaus Schulten. VMD – Visual Molecular Dynamics. Journal of Molecular Graphics, 14:33–38, 1996.
Florian Sittel, Abhinav Jain, and Gerhard Stock. Principal component analysis of molecular dynamics: On the use of cartesian vs. internal coordinates. The Journal of Chemical Physics, 141(1):07B605 1, 2014.
Vytautas Gapsys and Bert L. de Groot. Optimal superpositioning of flexible molecule ensembles. Biophysical Journal, 104(1):196–207, 2013.
Matthias Ernst, Florian Sittel, and Gerhard Stock. Contact- and distance-based principal component analysis of protein dynamics. The Journal of Chemical Physics, 143(24):244114, 2015.
Matthias Ernst, Steffen Wolf, and Gerhard Stock. Identification and validation of reaction coordinates describing protein functional motion: Hierarchical dynamics of t4 lysozyme. Journal of Chemical Theory and Computation, 13(10):5076–5088, 2017.
Florian Sittel, Thomas Filk, and Gerhard Stock. Principal component analysis on a torus: Theory and application to protein dynamics. The Journal of Chemical Physics, 147(24):244101, 2017.
Yuguang Mu, Phuong H. Nguyen, and Gerhard Stock. Energy landscape of a small peptide revealed by dihedral angle principal component analysis. Proteins: Structure, Function, and Bioinformatics, 58(1):45– 52, 2005.
Alexandros Altis, Moritz Otten, Phuong H. Nguyen, Rainer Hegger, and Gerhard Stock. Construction of the free energy landscape of biomolecules via dihedral angle principal component analysis. The Journal of Chemical Physics, 128(24):245102, 2008.
Florian Sittel and Gerhard Stock. Perspective: Identification of collective variables and metastable states of protein dynamics. The Journal of Chemical Physics, 149(15):150901, 2018.
Cattell RB. The scree test for the number of factors. Multivariate Behav Res, 1(2):245–76, 1966. PMID: 26828106.
Lucas Sawle and Kingshuk Ghosh. Convergence of molecular dynamics simulation of protein native states: Feasibility vs self-consistency dilemma. Journal of Chemical Theory and Computation, 12(2):861–869, 2016.
Mike Nemec and Daniel Hoffmann. Quantitative assessment of molecular dynamics sampling for flexible systems. Journal of Chemical Theory and Computation, 13(2):400–414, 2017.
Alan Grossfield and Daniel M. Zuckerman. Quantifying Uncertainty and Sampling Quality in Biomolecular Simulations, pages 23–48. Annual Reports in Computational Chemistry. Elsevier, 2009.
Rodrigo Cossio-Pérez, Juliana Palma, and Gustavo Pierdominici-Sottile. Consistent principal component modes from molecular dynamics simulations of proteins. Journal of Chemical Information and Modeling, 57(4):826–834, 2017. PMID: 28301154.
Andrea Amadei, Marc A Ceruso, and Alfredo Di Nola. On the convergence of the conformational coordinates basis set obtained by the essential dynamics analysis of proteins molecular dynamics simulations. Proteins: Structure, Function, and Bioinformatics, 36(4):419–424, 1999.
B. Hess. Convergence of sampling in protein simulations. Phys. Rev. E, 65:031910, Mar 2002.