La bioinformática y la investigación en las ciencias y tecnologías de la vida; una reseña sobre lo realizado en el Laboratorio de Ingeniería Genética y Biología Celular y Molecular
Bioinformatics and research in life sciences and technologies: a review of the work conducted at the Laboratory of Genetic Engineering and Cellular and Molecular Biology
Alejandro Simonin Carolina Susana CerrudoCristina Silvia Borio Damián Presti Fernando Maku Lassalle Franco Tomás Ledesma Franco Uriel Cuccovia WarletGonzalo Maciel Lucas Motta Lucas Ripoll Marchesano Wodniak LucasMarcos Bilen Mariano Nicolás BelaichLa bioinformática constituye actualmente un componente central de las ciencias y tecnologías de la vida, permitiendo integrar métodos computacionales, matemáticos y estadísticos para el análisis de grandes volúmenes de datos biológicos. En el presente trabajo se realiza una reseña sobre la evolución histórica de la bioinformática a través de los aportes y desarrollos realizados en el Laboratorio de Ingeniería Genética y Biología Celular y Molecular – Área Virosis de Insectos (LIGBCM-AVI) de la Universidad Nacional de Quilmes. Se describen los principales cambios conceptuales y tecnológicos ocurridos desde la era pregenómica, caracterizada por el estudio de genes individuales mediante secuenciación de Sanger, hasta la era posgenómica, impulsada por las tecnologías de secuenciación masiva y el análisis integrado de genomas, transcriptomas, proteomas y otros conjuntos de datos biológicos. Finalmente, se discute el impacto de la bioinformática en la investigación básica y aplicada del LIGBCM, su integración en la formación académica y los desafíos futuros asociados al desarrollo de estrategias multi-ómicas, inteligencia artificial y biología de sistemas.
Bioinformatics has become a central component of life sciences and technologies, enabling the integration of computational, mathematical, and statistical methods for the analysis of large volumes of data. In the present work, a review of the historical evolution of bioinformatics is presented through the contributions and developments carried out at the Laboratory of Genetic Engineering and Cellular and Molecular Biology – Insect Virosis Area (LIGBCM-AVI) of the Quilmes National University. The main conceptual and technological changes that occurred from the pregonomic era, characterized by the study of individual genes through Sanger sequencing, to the postgenomic era, driven by high-throughput sequencing technologies and the integrated analysis of genomes, transcriptomes, proteomes, and other biological datasets, are described. Finally, the impact of bioinformatics on LIGBCM basic and applied research, its integration into academic training, and future challenges associated with the development of multi-omics strategies, artificial intelligence, and systems biology are discussed.
Las ciencias de la vida deben manejar una inmensa cantidad de datos que no pueden ser interpretados sin ayuda computacional. Por esa razón, ha adquirido relevancia un enfoque científico interdisciplinario conocido como bioinformática, que tiene como objetivo resolver problemas biológicos complejos. Entonces, ¿qué es la bioinformática? Una respuesta sencilla podría ser que es una ciencia que analiza datos biológicos, biofísicos y bioquímicos a través de la aplicación de métodos matemáticos, estadísticos y técnicas computacionales, con el fin de extraer la información relevante almacenada en las biomoléculas y los sistemas biológicos de estudio. Si bien en sus inicios era solo una herramienta que ayudaba a comprender características simples de secuencias nucleotídicas y aminoacídicas, en la actualidad es una parte integral de la investigación biológica, y se ha convertido en una ciencia. Hoy en día, se requieren enfoques computacionales innovadores y de alto rendimiento (tecnologías high-throughput) para posibilitar el manejo de grandes volúmenes de datos biológicos y lograr resultados de alto impacto en campos como biología, biofísica, genética molecular, agricultura, biotecnología, medicina y en cualquiera de las disciplinas que conforman las ciencias y tecnologías de la vida (Figura 1). Es importante señalar que la bioinformática no es una ciencia experimental en el sentido tradicional, sino una ciencia predictiva que emplea los conocimientos derivados de la biología molecular para generar modelos y realizar análisis teórico-computacionales que permitan transformar los datos en información, y ésta, en conocimiento. No resulta extraño, en este sentido, que las y los primeros bioinformáticos (aunque sin título específico) pertenecieran a áreas biológicas, bioquímicas o de la salud, ya que la bioinformática se originó a partir de una necesidad específica de estas ciencias. Esta capacidad predictiva se traduce, sin embargo, en distintos modos de trabajo, según la etapa del proceso bioinformático de la que se trate. Por un lado, existen grupos de investigación abocados al desarrollo de métodos computacionales, cuya validación —realizada a partir de datos preexistentes— es numérica e interna y no requiere el diseño de nuevos experimentos. Por otro lado, cuando esos métodos se aplican para estudiar nuevos escenarios biológicos, los datos obtenidos mediante la experimentación alimentan a la bioinformática, la cual los resume, elimina redundancias, los analiza y extrae información, lo que permite elaborar nuevas hipótesis de trabajo y derivar en predicciones concretas. En este caso, la validación de dichas predicciones sí resulta necesaria y dependerá de estudios experimentales realizados por otros grupos o disciplinas, o por el mismo equipo si trabaja de forma integral, retroalimentando así a las ciencias biológicas. Por último, la bioinformática es una ciencia muy dinámica que se relaciona estrechamente con las diversas técnicas empleadas para obtener los datos biológicos y con sus respectivos avances. Su alto dinamismo se debe a que, a medida que se desarrollan nuevas técnicas experimentales, se generan nuevos tipos de datos biológicos que deben ir acompañados de desarrollos bioinformáticos capaces de manipularlos. Como ejemplo, puede mencionarse uno de los hitos de la biología molecular: la técnica de secuenciación de Sanger, reportada en 1977 [Sanger et al., 1977], que impulsó la creación de las primeras bases de datos de secuencias nucleotídicas como la EMBL-EBI Data Library (European Molecular Biology Laboratory – European Bioinformatics Institute) en 1980 [Hamm et al., 1986] y GenBank del NCBI (National Center for Biotechnology Information) en 1982 [Bilofsky et al., 1986], los cuales a su vez impulsaron el desarrollo de programas de comparación de secuencias como el Blast [Altschul et al., 1997] que, utilizando algoritmos de comparación de a pares y heurística, permiten identificar secuencias similares en una base de datos en tiempos computacionales razonables. Estos avances bioinformáticos surgieron en respuesta a las necesidades biológicas y no habían sido necesarios hasta ese momento.

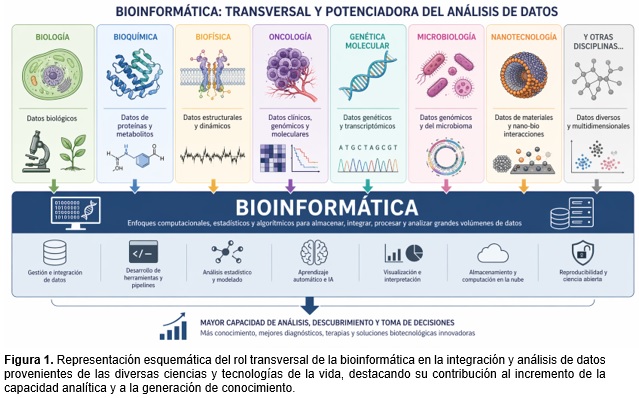
La investigación y el desarrollo en bioinformática abarcan desde la abstracción de las propiedades de un sistema biológico en modelos computacionales hasta el desarrollo y la implementación de nuevos algoritmos para el análisis de datos, su transformación en información, y el desarrollo de bases de datos y herramientas web para facilitar su acceso a la comunidad científica. La estrecha vinculación entre investigación y enseñanza, propia de la Universidad Nacional de Quilmes (UNQ), explica que la bioinformática se haya incorporado no solo a sus áreas de investigación, sino también a sus aulas. Al ser entendida como una ciencia de la información, y considerando su capacidad de potenciar el alcance de otras disciplinas científicas, se ha generado un nuevo paradigma educativo en el que se debe garantizar una formación integrada en biología e informática suficiente para satisfacer la demanda de un nuevo perfil profesional: técnicas/os, licenciadas/os o maestrandas/os en bioinformática. Estos deben comprender tanto el lenguaje biológico como computacional, y su función principal será el diseño y aplicación de herramientas computacionales para resolver problemas biológicos específicos. Actualmente, la UNQ cuenta con una carrera de grado (Licenciatura en Bioinformática) y una de posgrado (Maestría en Bioinformática y Biología de Sistemas) relacionadas con esta disciplina. Inclusive, el Doctorado en Ciencia y Tecnología acepta tesis exclusivamente bioinformáticas, evidenciando la posibilidad de desarrollar una trayectoria completa en bioinformática a lo largo de todos los niveles académicos. Asimismo, la mayoría de los laboratorios de investigación en el Departamento de Ciencia y Tecnología de la UNQ aplican en algún momento del desarrollo de sus líneas de investigación conocimientos bioinformáticos, ya sea en el análisis previo al desarrollo experimental (para simplificarlo o volverlo más específico), la generación de bases de datos (para poder almacenar de forma adecuada los datos experimentales generados), en el análisis de los datos (para poder obtener la información almacenada en los mismos), o incluso desarrollando algoritmos específicos para poder analizar los datos generados. En particular, el Laboratorio de Ingeniería Genética y Biología Celular y Molecular – Área Virosis de Insectos (LIGBCM-AVI), dirigido actualmente por los Doctores Mariano Nicolás Belaich y Marcos Fabián Bilen, es un grupo de trabajo consolidado en investigación básica y aplicada sobre los baculovirus, flavivirus y otros entomopatógenos, con áreas de aplicación muy diversas: control biológico de plagas agrícolas, terapias génicas para animales y humanos, big data biológica para desarrollar soluciones de precisión en salud y agricultura, sistemas de diagnóstico molecular, evolución de proteínas y servicios de genómica para la industria. Si bien el laboratorio cuenta con más antecedentes en áreas no vinculadas a la bioinformática, su desarrollo en este campo ha sido fuertemente influenciado por su fundador y exdirector, el Dr. P. Daniel Ghiringhelli. En este sentido, él dejó un importante legado académico, formando a numerosos investigadores e investigadoras en el área, y fue una pieza central en la creación de la Licenciatura en Bioinformática y la Maestría en Bioinformática y Biología de Sistemas. Debido a esto, casi todos los proyectos y publicaciones del LIGBCM-AVI incorporaron a lo largo del tiempo herramientas bioinformáticas, además de que en los últimos años se han desarrollado estudios íntegramente bioinformáticos.
En este contexto, en el presente manuscrito se realiza un recorrido por la historia de la bioinformática, de la mano de los aportes realizados por el LIGBCM-AVI de la UNQ (fundado en 1997 por el Dr. Ghiringhelli); con el objetivo de describir no sólo la evolución de dicha ciencia sino también su impacto en distintas investigaciones. En las siguientes secciones profundizaremos en la historia de la bioinformática y cómo sus aportes permitieron transformar datos en información útil para las investigaciones y desarrollos del LIGBCM-AVI.
La historia de la bioinformática, desde sus inicios (aún antes de su denominación formal) hasta la actualidad, puede dividirse en dos eras –la pregenómica y la posgenómica–, que se distinguen claramente por el enfoque utilizado y la magnitud de los datos manipulados (Figura 2A). En la etapa pregenómica, desde aproximadamente 1980 hasta 2004, los esfuerzos se concentraban en la caracterización de genes particulares (principalmente genes codificantes de enzimas) o secuencias con función biológica discreta; y las secuencias de ácidos nucleicos se obtenían principalmente por el método de secuenciación de Sanger [Sanger et al., 1977]. Si bien esta tecnología fue luego automatizada, presentaba una capacidad de lectura de entre 500-800 bases y costos elevados para regiones genómicas extensas. En resumen, la metodología consistía en la generación de una biblioteca genómica mediante la digestión parcial del genoma de interés con endonucleasas de restricción, el clonado en un vector plasmídico, la transformación en Escherichia coli seguido de la identificación de los clones de interés mediante hibridación con sondas nucleotídicas diseñadas ad hoc. Posteriormente, se procedía a secuenciar por Sanger de forma manual o automatizada. En otras palabras, se buscaba una secuencia desconocida con identidad conocida. En cuanto a los algoritmos bioinformáticos utilizados, los mismos consistían en alineamientos de a pares (para poder ensamblar las secuencias obtenidas), de búsqueda en bases de datos (para poder recuperar secuencias similares al gen, proteína o genoma secuenciado), de alineamientos múltiples de secuencias aminoacídicas o nucleotídicas (para poder comparar un conjunto de secuencias y determinar regiones conservadas), algoritmos de inferencia filogenética (para determinar la relación evolutiva del gen novedoso con los otros genes previamente caracterizados), algoritmos de predicción de caracterización de proteínas (para poder estudiar la estructura primaria, secundaria y terciaria) y algoritmos de inferencia de función, entre otros. Como se mencionó anteriormente, los avances en el conocimiento molecular generan desafíos en todos los niveles de la bioinformática, que requiere una constante adaptación e innovación para mantenerse actualizada. Esto fue lo que ocurrió aproximadamente en 2004 cuando comenzó la era posgenómica, con el advenimiento de las tecnologías de secuenciación masiva de nueva generación o NGS (Next Generation Sequencing, también llamada luego secuenciación de segunda generación), y se extiende hasta la actualidad. El avance tecnológico fue tal que provocó el cambio de paradigma y marcó el fin de la era pregenómica. Tecnologías como Roche 454 (pirosecuenciación) [Margulies et al., 2005], Illumina (secuenciación por síntesis) [http://www.illumina.com], Applied Biosystem’s SOLiD (secuenciación por ligación) e Ion Torrent (secuenciación por síntesis) [https://www.thermofisher.com], facilitaron significativamente la obtención de secuencias genómicas completas con reducciones sustanciales en tiempo y costo gracias a la paralelización de la obtención de los datos, ya que son tecnologías de high-throughput. Si bien la longitud de cada lectura individual es inferior a las del método de Sanger, su baja tasa de error (también respecto a la tecnología Sanger, técnica de referencia de ese momento), más el gran número de datos simultáneos y las mejoras en los procedimientos computacionales, revolucionaron de modo significativo la adquisición de secuencias genómicas durante este siglo. Este cambio tecnológico exigió, a su vez, una adaptación significativa de las herramientas y los métodos bioinformáticos hasta entonces disponibles. Mientras que en la era pregenómica se construía desde lo micro (genes, bricks génicos y otros elementos genómicos) hacia lo macro (genomas), en la era posgenómica se invierte esta premisa, primero obteniendo el genoma completo y luego determinando la función biológica de los datos obtenidos (Figura 2A). En 2011, PacBio [http://www.pacb.com] innovó en las NGS con el desarrollo de una metodología de secuenciación de lecturas largas, que posibilitó obtener la secuencia de una sola molécula de ADN en tiempo real (Single-Molecule Real-Time sequencing technology), eliminando las variantes de PCR (Polymerase Chain Reaction) que formaban parte del procedimiento previo a la secuenciación, dando lugar así a la tercera generación de tecnologías de secuenciación. Posteriormente, en 2014, surge Nanopore [https://www.nanoporetech.com] como otra tecnología de tercera generación, con un enfoque muy distinto, pero manteniendo la característica clave de secuenciar moléculas únicas. Si bien ambas tecnologías presentan un menor rendimiento de datos que las de segunda generación, y una tasa de error un tanto mayor (aunque mejorando en este aspecto en los últimos años), sus lecturas de varios miles de nucleótidos posibilitaron cubrir regiones genómicas difíciles de secuenciar y/o ensamblar, habilitando obtener datos genómicos de extremo a extremo de cada cromosoma.

Las tecnologías de NGS incrementaron exponencialmente la disponibilidad de fragmentos de secuencias genómicas, lo que impulsó la evolución de la bioinformática para automatizar el ensamblado de estos en genomas completos [MacCannell, 2019]. Actualmente, ya no se trabajaba con algunas pocas secuencias para ensamblar, sino con miles de millones (cortas o largas, según la tecnología empleada) que debían compararse con un genoma conocido para realizar un ensamblado con referencia (mediante nuevos algoritmos de alineamiento desarrollados específicamente para tal fin); ó compararse entre sí para realizar un ensamblado de novo (mediante algoritmos de alineamientos nuevos o readaptados), cuando no se disponía de una referencia. Asimismo, se desarrollaron herramientas capaces de caracterizar los genomas completos obtenidos y de abordar datos provenientes de tecnologías de transcriptómica, proteómica y genómica funcional de alto rendimiento; mejorando considerablemente la identificación de los genes junto con su anotación estructural y funcional. Esto incluyó el estudio de aspectos estáticos, como la identificación de genes y proteínas; y dinámicos, como los procesos de transcripción y traducción, las modificaciones postraduccionales y las interacciones proteína-proteína. Es importante también mencionar que los algoritmos desarrollados en la era pregenómica siguen siendo utilizados en la actualidad, aunque ya no se aplican a la caracterización de genes o proteínas individuales, sino al análisis de todos los genes codificados en genomas ensamblados, y en algunos casos han sido adaptados para manejar la gran cantidad de secuencias proteicas o nucleotídicas con las que se trabaja. Las tecnologías de las tres generaciones siguen siendo utilizadas actualmente para obtener distintos conjuntos de datos biológicos, cada una con sus ventajas y desventajas. Entre sus aplicaciones más relevantes, se pueden mencionar: la secuenciación y el ensamblado de genomas completos –o whole genome sequencing– de novo o con referencia, aportando a las esferas de la genómica y la epigenética; las secuenciaciones dirigidas a zonas específicas, con el objetivo de estudiar variabilidad alélica o emplear metagenómica para estimar poblaciones de organismos en un contexto dado, entre otros; y la secuenciación de transcriptomas –o RNA-seq, en todas sus variantes– para análisis fenotípicos de organismos, tejidos, o poblaciones celulares [Ibañez-Lligoña et al., 2023].
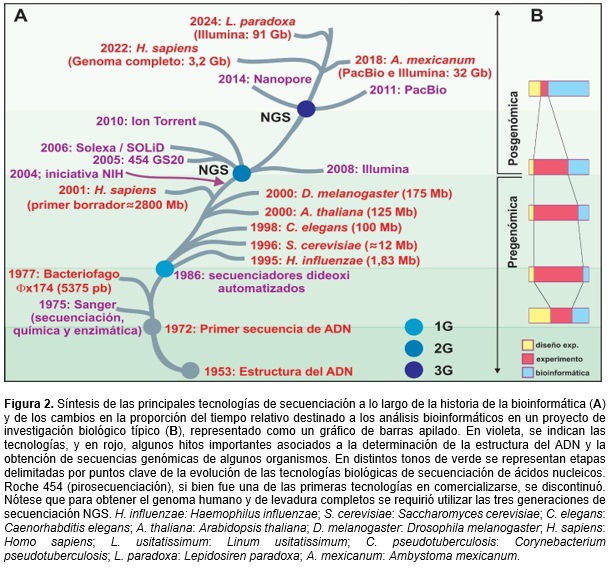
Por otro lado, resulta relevante analizar cómo ha variado, con el correr de los años, el tiempo dedicado a cada parte de un proyecto realizado en un laboratorio de investigación en ciencias y tecnologías de la vida (Figura 2B). En los inicios de la biología molecular, cada proyecto se dividía en 3 grandes momentos, con duraciones similares: diseño experimental, experimento y análisis de los datos. Posteriormente, en la época de oro de la biología molecular, la etapa más demandante era el experimento, ya que había muchas técnicas novedosas y factores para poner a punto. Sin embargo, con la aparición de la secuenciación profunda, el tiempo dedicado al desarrollo del experimento es mucho menor y la etapa más demandante es la asociada al análisis de los datos (de la cuál es altamente responsable la bioinformática).
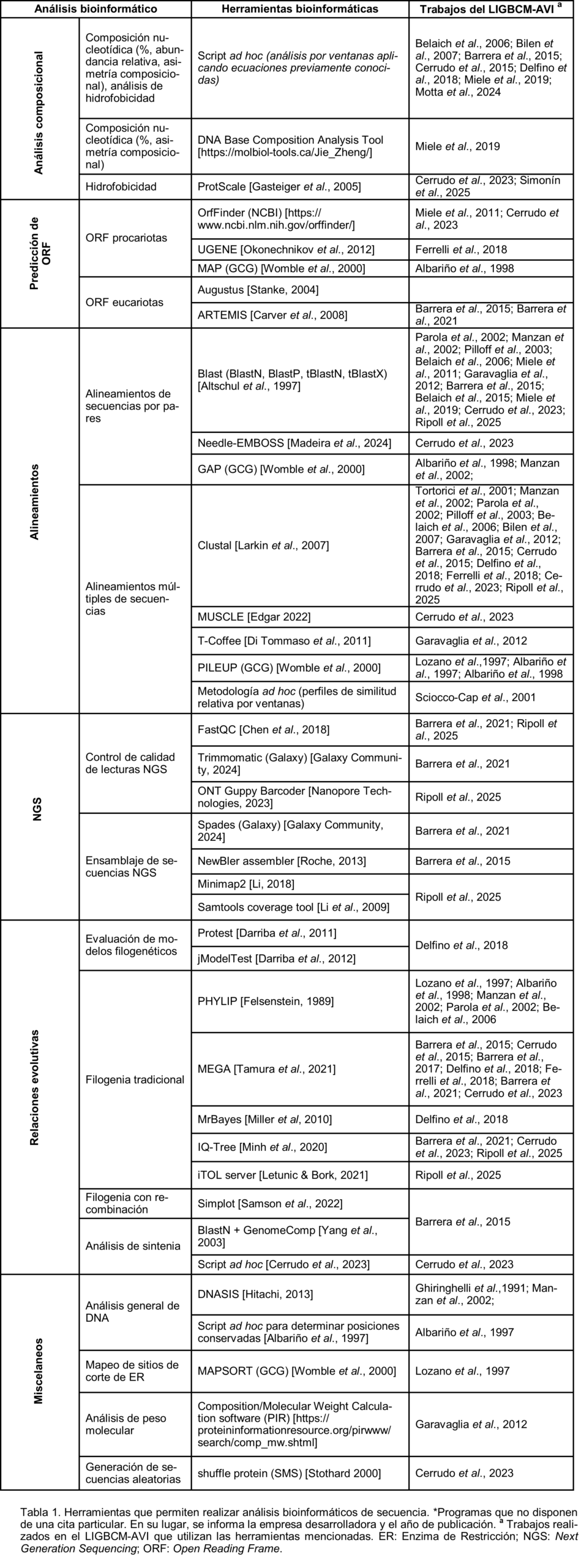
Si bien hasta ahora diferenciamos entre los análisis bioinformáticos utilizados y desarrollados en las eras pre y posgenómicas, podemos también pensar en diferentes clasificaciones de los análisis bioinformáticos que serían transversales a las dos eras previamente definidas. Por ejemplo, sólo para mencionar algunas, podría plantearse una clasificación según su área de aplicación (desarrollo de bases de datos y repositorios, biología computacional, biología molecular, biotecnología, biología de sistemas, clínica ó incluso farmacogenómica y medicina personalizada), según el tipo de macromolécula que estudian (ADN, ARN, proteínas), según el flujo de trabajo computacional utilizado (desarrollando algoritmos, utilizando plataformas o servidores preexistentes, utilizando diferentes sistemas operativos o lenguajes de programación), ó según la característica de la macromolécula que podemos estudiar, es decir la información que genera ese análisis. A continuación, presentaremos algunas herramientas o análisis bioinformáticos agrupados en función de la última clasificación mencionada, ya que es la que mejor se adapta a los ejes de trabajo del laboratorio. En este sentido, podemos distinguir tres categorías, los análisis para caracterizar las secuencias, las estructuras, o la función de las macromoléculas; todas ellas en conjunto permiten realizar una caracterización completa de un sistema biológico de interés (a nivel de secuencias, estructuras y/o función). Asimismo, la bioinformática cuenta con aspectos inherentes, independientes de la característica biológica estudiada o del tipo de información generada. Como ejemplo, podemos mencionar la estadística, la programación, la inteligencia artificial y el desarrollo de bases de datos, disciplinas que serán aplicados transversalmente en las tres categorías mencionadas (secuencia, estructura y función). Para evaluar y validar los resultados bioinformáticos que, por naturaleza, son predictivos, se deben emplear métodos estadísticos. A su vez, se puede hacer uso de la programación para desarrollar herramientas o programas que automaticen la obtención de información biológica, así como flujos de trabajo continuos, estructurados y ordenados de tareas, procesos o herramientas, diseñados para analizar datos biológicos (pipelines), o también para el desarrollo de bases de datos específicas del tipo de información generada por cada herramienta. Además, estas pueden verse potenciadas mediante el uso de la inteligencia artificial y el machine learning, el cual permite que los algoritmos aprendan de los datos biológicos, identifiquen patrones y realicen predicciones cada vez más precisas.
Debido al dinamismo de la bioinformática y la variedad de algoritmos y programas disponibles, no resulta práctico describir los programas en detalle. En su lugar, en los siguientes apartados se presentará un breve resumen de los análisis bioinformáticos que se pueden emplear para transformar datos en información, obteniendo la información secuencial, estructural o funcional de las macromoléculas. A su vez, cada apartado se nutrirá de tablas que mencionan programas o herramientas disponibles para realizar los análisis, con el objetivo de que el lector conozca algunas de las más utilizadas, así como los trabajos del LIGBCM-AVI en los que fueron aplicadas, de modo que pueda acceder a bibliografía específica donde dichas herramientas han sido empleadas.
Caracterización de secuencias. Los ácidos nucleicos y las proteínas pueden ser caracterizados analizando su composición nucleotídica/aminoacídica [Moeckel et al., 2023]. Por ejemplo, al calcular el porcentaje de nucleótidos G y C (%GC) de una molécula de ADN, se puede estimar la estabilidad de la doble hebra. Cuanto mayor sea este porcentaje, mayor será la energía necesaria para romper los puentes de hidrógeno que las mantiene unidas. Este cálculo también es útil para identificar la taxonomía de secuencias incógnitas o para detectar inserciones de genes o secuencias provenientes de otros entes biológicos, ya que el %GC es característico de cada organismo y su valor no suele variar significativamente a lo largo de su genoma. Otra caracterización composicional que puede realizarse sobre secuencias nucleotídicas es la distribución de oligonucleótidos de corta longitud o repeticiones, que suele relacionarse directamente con detalles estructurales asociados a funciones particulares [Lo et al., 2023]. En cuanto a las proteínas, la caracterización composicional puede utilizarse para identificar su hidrofobicidad e hidrofilicidad general y local, en regiones o dominios particulares. Estos análisis suelen combinarse con estudios estructurales, ya que las zonas más expuestas al solvente suelen presentar una composición mayormente hidrofílica, mientras que las zonas ocultas suelen ser más hidrofóbicas (Tabla 1). Por otro lado, las secuencias de aminoácidos o nucleótidos pueden compararse a través de la búsqueda de similitud utilizando diferentes estrategias de alineamientos [Zhang et al., 2024; Ou et al., 2023]. En este sentido, se puede realizar la comparación de dos o más secuencias –alineamientos de a pares o múltiples– para detectar regiones o subsecuencias de composición igual o equivalente –regiones de similitud–. A lo largo del tiempo, surgieron diferentes estrategias y algoritmos de alineamiento que permiten resolver diversas preguntas biológicas. Por ejemplo, si se dispone de una secuencia novedosa y se desea identificar secuencias similares en bases de datos, o si se comparan dos secuencias para evaluar la presencia de regiones de similitud, o bien si se analiza un conjunto de secuencias relacionadas para determinar regiones conservadas y variables, o incluso si se cuenta con una colección de secuencias y se busca generar un perfil flexible capaz de detectar nuevos miembros, estos problemas pueden abordarse mediante estrategias de alineamiento tradicionales, ya sean de a pares o múltiples, locales o globales [Zhai et al., 2025]. Sin embargo, en la era posgenómica surgieron nuevos algoritmos de alineamiento para poder alinear miles de millones de datos crudos de secuencias NGS correspondientes a un genoma y concatenarlos adecuadamente para reconstruir la secuencia genómica (en el caso de ensamblaje de novo) o poder alinear las miles de millones de lecturas contra un genoma particular para generar el ensamblado (en el caso de contar con una referencia), así sean secuencias de lecturas cortas [Kulkarni et al., 2017] o largas [Van Almsick et al., 2026]. Otro de los análisis que se puede realizar, basándose en la información biológica contenida en las secuencias de las macromoléculas, es una inferencia filogenética utilizando un conjunto de secuencias relacionadas. Este análisis, fuertemente ligado a los alineamientos múltiples globales de secuencias (MSA), permite determinar la relación de las secuencias en función de su historia evolutiva; es decir, describir cómo los miembros de esa familia de secuencias podrían haber derivado durante la evolución [Redelings et al., 2024]. Los distintos algoritmos filogenéticos que existen permiten predecir la historia evolutiva de secuencias nucleotídicas y aminoacídicas mediante el trazado de un camino desde el ancestro común hacia las distintas ramificaciones que se fueron generando a partir de las variaciones de secuencia. Un descubrimiento fundamental de los estudios evolutivos fue que organismos de taxonomías muy lejanas emplean sets de genes similares para funciones biológicas básicas. Un modelo evolutivo bastante aceptado asegura que estos genes han sido alterados, copiados y rearreglados, a partir de un set básico original –perteneciente a un ancestro común muy lejano–, para crear nuevos genes y familias de genes que hoy en día se pueden encontrar en los distintos organismos. La variación evolutiva de las secuencias puede representarse en un gráfico en forma de árbol –un árbol filogenético–, en el cual se podrán distinguir clados (agrupamientos de taxas) y observar distancias evolutivas. Descubrir el arreglo de las ramas y la longitud que mejor represente las relaciones entre las secuencias será el objetivo de la inferencia filogenética. Los árboles reflejaran la cantidad de cambios y las relaciones ancestrales entre las secuencias, pudiendo revelar qué genes han seguido la misma ruta evolutiva y cuáles no. Para esto, en la actualidad existe una gran cantidad de modelos evolutivos y métodos de análisis distintos, y también una gran variedad de programas que implementan alguno o varios de estos algoritmos para analizar, de diversas maneras, las diferencias entre los caracteres de las secuencias de un MSA [Jacques et al., 2023]. Incluso existen algoritmos que permiten hacer inferencia filogenética sin alineamientos, que adquieren relevancia al necesitar comparar genomas grandes [Sims et al., 2009].

A su vez, como los genomas pueden sufrir recombinaciones y otras modificaciones estructurales, pueden generarse secuencias que representan datos conflictivos al ser analizados por los métodos de filogenia tradicional, y deben emplearse algoritmos capaces de resolver estos problemas [Samson et al., 2022]. Una vez realizado el análisis filogenético, se puede determinar si algunas de las secuencias son homólogas, es decir, tienen un ancestro en común y son similares, o presentan homoplasía: una similitud que no se debe a un ancestro común, sino que puede surgir por evolución independiente (convergencia o paralelismo) o por la recuperación de un estado ancestral (reversión). Dentro del conjunto de secuencias homólogas se pueden distinguir las secuencias ortólogas (existen en diferentes especies, pero provienen de un gen ancestral común y retienen la misma función) y parálogas (genes homólogos que divergieron después de un evento de duplicación génica dentro de un genoma, que pueden presentar funciones diferentes pero relacionadas como los ARNt). El concepto de ortología, junto con los algoritmos disponibles para la detección de ortólogos, adquiere mayor relevancia al considerar ortólogos remotos. Este tipo de análisis permite identificar secuencias con relaciones evolutivas distantes y facilitan la inferencia de función [Hsieh et al., 2025].
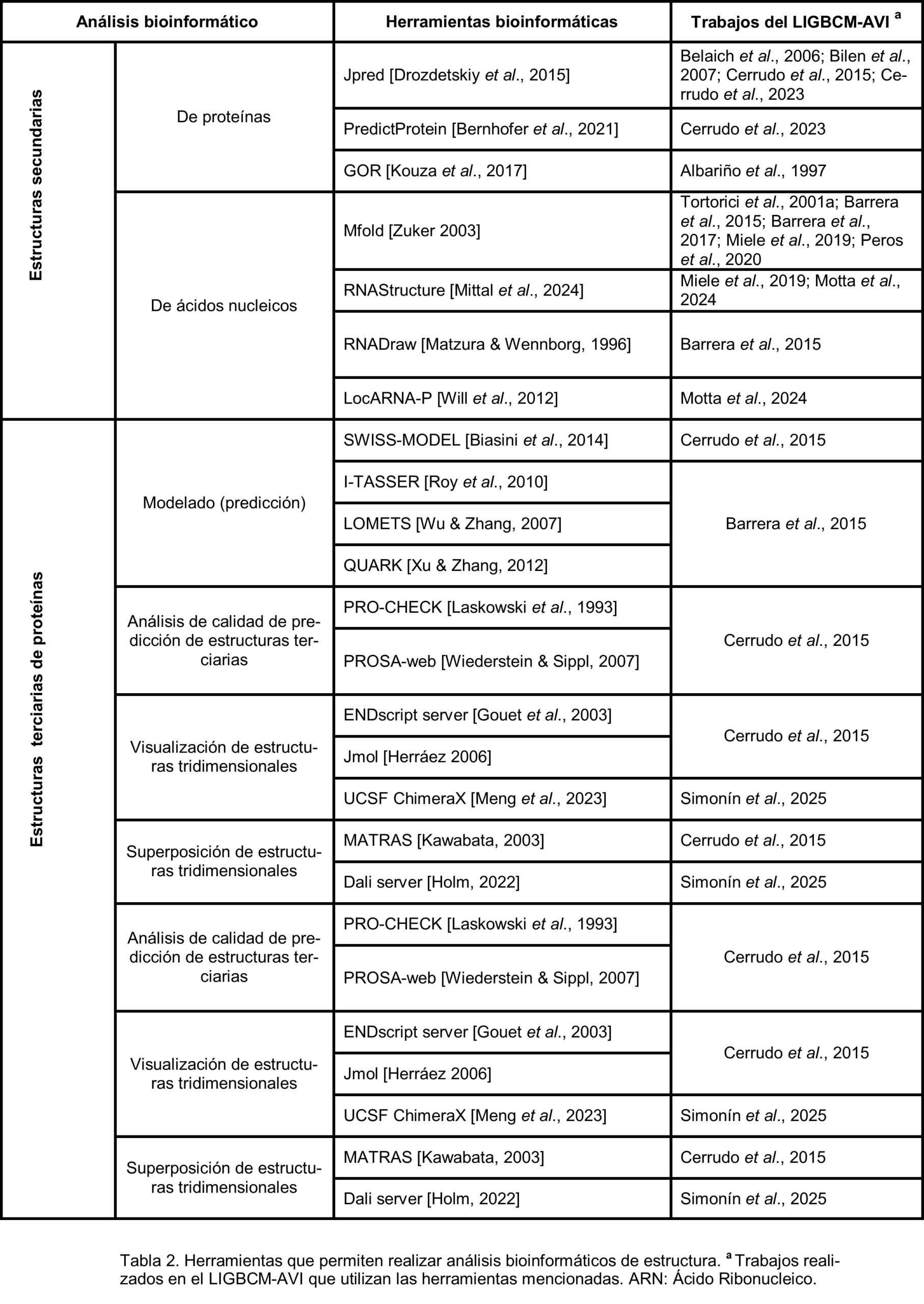
Caracterización de estructura. En la actualidad es posible predecir la estructura secundaria, terciaria e incluso cuaternaria de moléculas de ARN, ADN, proteínas y estructuras híbridas como los ribosomas (Tabla 2). Entre estas macromoléculas, la estructura del ADN es la más simple, ya que consiste, en la gran mayoría de los casos, en una doble hebra complementaria e hibridizada, cuya principal variación estructural corresponde a sus conformaciones B-, A- o Z-ADN. De este modo, la caracterización estructural de ADN más relevante es la predicción de sistemas híbridos dónde dicha macromolécula se encuentra interactuando con proteínas de unión a ADN. Por otro lado, los primeros programas de caracterización estructural fueron desarrollados para la predicción de la estructura secundaria de ARN simple cadena [Sato & Hamada, 2023], ya que sus secuencias son relativamente cortas y su función se encuentra estrechamente relacionada con su plegamiento. Las proteínas son las macromoléculas que presentan mayor complejidad estructural y, por lo tanto, mayor dificultad para realizar predicciones acertadas.

Uno de los grandessueños de la bioinformática fue poder predecir las estructuras secundaria y terciaria de una proteína a partir de la estructura primaria. Si bien a partir del surgimiento de AlphaFold [Abramson et al., 2024] esto parece cada vez más factible, hay que tener en cuenta que dentro de la célula pueden darse procesos de clivaje o modificaciones químicas post-traduccionales, además de proteínas chaperonas que, junto con las membranas celulares, pueden asistir a las proteínas en su proceso de plegamiento. La bioinformática puede ayudarnos mediante diversos programas, no solo a predecir estructuras proteicas sino también a caracterizar y obtener información de las estructuras [Kuzu et al., 2025], además de poder compararlas para determinar si existen regiones similares entre ellas [Alanazi et al., 2025].
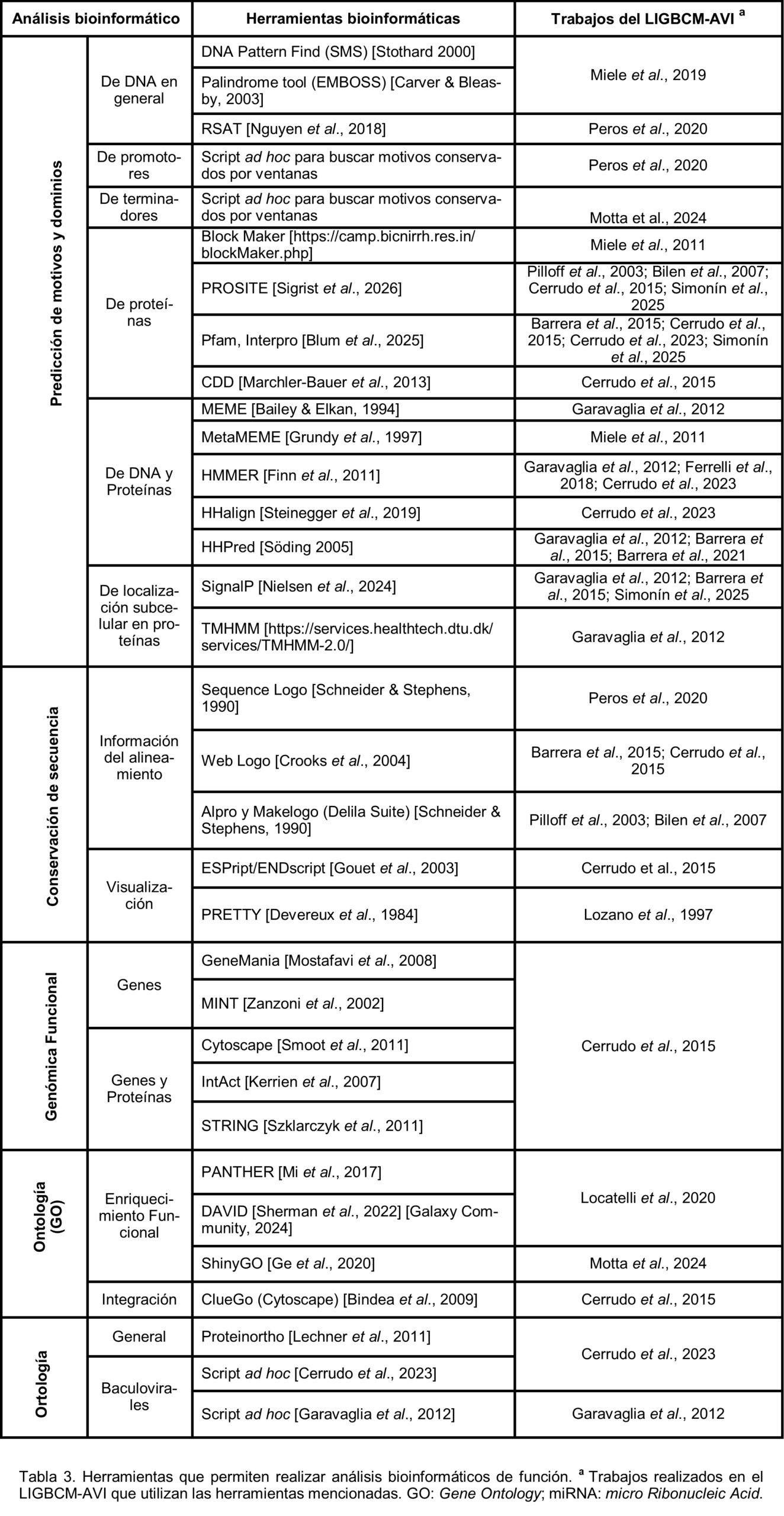
Caracterización de función. Para la predicción de la función de las macromoléculas (Tabla 3), pueden analizarse las similitudes de secuencia a fin de identificar regiones conservadas que permitan inferir motivos o dominios. La búsqueda de motivos, dominios y familias proteicas puede ser útil para caracterizar secuencias nucleotídicas y/o aminoacídicas (predicción de regiones promotoras, regulatorias o de unión de factores transcripcionales en genomas, predicción de función o clasificación de familias en proteomas). Además, pueden servir para detectar o recuperar secuencias homólogas remotas. Estos se basan en métodos de comparación de secuencias más sofisticados que los MSA mencionados anteriormente, incluyendo a los algoritmos generadores de patrones sintácticos, perfiles o matrices de peso, como las matrices de puntuación específica de posición [Sigrist et al., 2026]; y los modelos ocultos de Markov o HMMs por Hidden Markov Models [Blum et al., 2025]. También podemos analizar datos de transcriptómica y proteómica para evaluar si un conjunto de genes se expresa en determinado momento, si modifica su expresión frente a una condición específica, o si se sintetizan proteínas de interés [Amarapathy et al., 2026]. Además, podemos realizar análisis de ontología (GO, por Gene Ontology) para transformar listas de genes en conocimiento biológico a través de la asignación de un conjunto de términos controlados que describen la función, el proceso biológico o el componente celular de un producto génico en base a la anotación genética entre especies [Ilgisonis et al., 2022].

Asimismo, el campo de la genómica funcional utiliza los datos producidos por proyectos genómicos, transcriptómicos y proteómicos para describir funciones e interacciones de genes y de proteínas, analizando los aspectos dinámicos de un gen, en lugar de los aspectos estáticos de la información genómica (como la secuencia o estructura). Estos análisis involucran, por lo tanto, muchas de las estrategias bioinformáticas y experimentales ya mencionadas, para dilucidar la función del genoma en relación con el fenotipo; es decir, permite caracterizar medidas de actividad molecular, la organización, el control de las rutas genéticas y las interacciones entre macromoléculas; lo que en conjunto hace a la fisiología de un organismo [Lam et al., 2017]. Por último, a partir de los análisis de ortología mencionados previamente podemos también inferir función de proteínas [Majidian et al., 2025].
Si bien, como mencionamos antes, el LIGBCM-AVI surge como un laboratorio de biología molecular y biotecnología dedicado a la ingenierización de baculovirus para mejorar sus aplicaciones, casi desde sus inicios se realizaron análisis bioinformáticos en las diferentes líneas de trabajo. Esto implicó que se siguiera avanzando en el estudio de esta ciencia y que, con el correr de los años, adquiriera cada vez más importancia, instaurando líneas completas de bioinformática. Para mostrar el recorrido del LIGBCM de la mano de la bioinformática, se narrará a continuación los trabajos desarrollados en el mismo, que contaron con análisis bioinformáticos. Para ello, se realizó una búsqueda bibliográfica exhaustiva utilizando PubMed (https://https://pubmed.ncbi.nlm.nih.gov/) con las siguientes palabras claves: Ghiringhelli PD[Author] OR Belaich MN[Author] OR Bilen MF[Author]; lo que permitió obtener 78 publicaciones entre 1989 y 2026. A partir de estos resultados, se realizó un filtro inicial por fecha (año 1998), para poder conservar los resultados posteriores a la creación del Laboratorio (recuperando 70 trabajos) y finalmente se revisaron las publicaciones seleccionando las que tuvieran análisis bioinformáticos realizados, buscando el detalle de las herramientas utilizadas para tal fin. A continuación, se presenta una reseña, diferenciando entre la era pregenómica y la posgenómica.
Durante la era pregenómica. En la era pregenómica de la bioinformática, con la participación (en diferentes posiciones de autoría) del Dr. Ghiringhelli, se publicaron 10 trabajos de investigación en revistas internacionales en los que se realizaron análisis similares a los descritos para esta era. De estos, los primeros seis corresponden a la caracterización de Arenavirus (virus transmitidos principalmente por roedores cuyo genoma está compuesto de ARN segmentado, consistiendo de un segmento largo y otro corto, L y S respectivamente), mientras que los siguientes cuatro corresponden a Baculovirus (familia de virus infectivos para artrópodos cuyo genoma está compuesto de ADN circular doble cadena de entre 80-180 kilopares de bases). Los estudios de Arenavirus incluyen la caracterización del fragmento genómico S (que codifica para las proteínas N y GPC) de la cepa MC2 de Mammarenavirus juninense (JUNV) [Ghiringhelli et al., 1991], la misma caracterización para las cepas Candid#1 (cepa atenuada, utilizada como componente activo de una vacuna contra la Fiebre Hemorrágica Argentina), XJ#44 y XJ (el prototipo de la especie) [Albariño et al., 1997]; en estos casos se obtuvo la secuencia completa del segmento S mediante clonado de los fragmentos del ARN S, se realizó un ensamblado por alineamiento de los fragmentos, la predicción de los genes codificados en el mismo, se predijo la estructura secundaria de las regiones no codificantes y se compararon las secuencias aminoacídicas predichas (proteínas N y GPC) con las conocidas previamente. A su vez, se trabajó en el diseño de cebadores (o primers, secuencias cortas de ácidos nucleicos -de entre 18 a 25 nucleótidos- que sirven como punto de inicio para la síntesis de una nueva cadena de ADN) específicos de Arenavirus para la amplificación de regiones conservadas del fragmento S de cualquier especie (tanto las del viejo como las del nuevo mundo), utilizando las secuencias disponibles en las bases de datos, programas de MSA y de filogenia [Lozano et al., 1997]; en el estudio filogenético de Arenavirus basado en las secuencias nucleotídicas del fragmento genómico S y las secuencias aminoacídicas de las proteínas N y GPC [Albariño et al., 1998]; y en la caracterización de la proteína N de la cepa MC2 de JUNV [Tortorici et al., 2001a; Tortorici et al., 2001b], mediante predicción de estructuras secundarias de los extremos del ARN S y MSA de la secuencia aminoacídica de la proteína N y búsqueda de motivos. Por otra parte, los primeros tres estudios de Baculovirus se enfocaron en un Granulovirus (EpapGV) aislado de larvas de Epinotia aporema. En estos se realizaron caracterizaciones biológicas, morfológicas y bioquímicas de los viriones [Sciocco-Cap et al., 2001], se obtuvieron mapas físicos y genéticos [Parola et al., 2002] y se caracterizó el gen de la proteína egt (ecdysteroid UDP-glycosyltransferase) [Manzán et al., 2002]; para ello se obtuvo la secuencia aminoacídica terminal directa de la proteína Granulina y se comparó con las depositadas en la base de datos mediante MSA y consensos, se predijeron ORF (por Open Reading Frame en inglés, marcos de lectura abiertos) sobre los fragmentos de secuencias obtenidos, se buscaron secuencias similares con el Blast y se realizaron análisis filogenéticos y perfiles de similitud relativa de aminoácidos. El cuarto y último estudio Baculoviral analizó la secuencia codificante del gen de la proteína GP64 del Nucleopolihedrovirus AgMNPV (Anticarsia gemmatalis Multiple Nucleopolyhedrovirus), cepa de Santa Fe (SF), en donde a partir de los fragmentos secuenciados se obtuvo el gen gp64 y se caracterizó realizando búsqueda de motivos regulatorios (promotores y poliadenilación), búsqueda en bases de datos de secuencias aminoacídicas y nucleotídicas similares, MSA y perfiles de similitud relativa [Pilloff et al., 2003].
Durante la era posgenómica. En la era posgenómica, por lo general, se parte de la construcción de genomas completos y la posterior transformación de los datos en información (por ejemplo, ubicación de genes particulares y asignaciones de identidad). En el marco de los proyectos desarrollados en el LIGBCM durante la etapa inicial de la era posgenómica (2004-2012), se llevó a cabo la caracterización biológica de la infección de larvas de Anticarsia gemmatalis con aislamientos baculovirales de Argentina y Brasil (AgMNPV-SF y AgMNPV-2D, respectivamente), realizando estudios genómicos comparativos de dichos virus y estudiando por microscopía óptica y electrónica sus dos fenotipos (BV y ODV; Budded Virions y Occlusion-Derived Virions). Además, se obtuvieron las secuencias de diversos genes del virus AgMNPV-SF, se realizaron análisis transcriptómicos de alguno de ellos (ie-1, gp64, p74, p10 y poliedrina) durante el curso de infecciones en células UFL-Ag-286 con el mismo virus, se expresaron cuatro proteínas recombinantes (IE-1, GP64, P74 y Poliedrina) en sistemas procariotas y eucariotas, y se generaron sueros policlonales contra muchas de ellas. También, se evaluó la amplificación in vitro de genomas baculovirales usando la ADN polimerasa del bacteriofago Φ29, se optimizó una metodología de sincronización celular previo a la infección con baculovirus, se realizaron estudios proteómicos del sistema virus-hospedador, se caracterizaron parcialmente dos baculovirus de mariposas diurnas, y se obtuvieron nuevos elementos móviles recuperados de líneas celulares de insecto [Belaich et al., 2006; Bilen et al, 2007; Mengual Gómez et al., 2010; Rodríguez et al., 2011a; Rodríguez et al., 2011b; Ferrelli et al., 2011; Rodríguez et al., 2012; Ferrelli et al., 2012]. En estos trabajos, los análisis bioinformáticos implicaron la obtención de la secuencia de la región genómica o genoma completo, la caracterización de la región codificante (predicción de ORF) junto con las regiones regulatorias (análisis de motivos), estudios de sintenia, análisis filogenético de diferentes tipos y caracterización de proteínas con análisis de similitud, predicción de estructura secundaria, perfiles de hidrofobicidad y determinación de dominios o motivos funcionales. cDurante los siguientes años se han realizado en el laboratorio diversos estudios completamente bioinformáticos sobre distintos genomas previamente depositados en las bases de datos públicas, descripción de proteínas de interés y ensamblado de genomas utilizando secuenciación NGS tanto de segunda como de tercera generación, desarrollándose incluso algunas aplicaciones y algoritmos para llevar a cabo dicha caracterización. En lo que respecta al análisis bioinformático sobre genomas baculovirales, estos estuvieron inicialmente centrados en la determinación de ortologías. El análisis de los 58 genomas baculovirales conocidos en 2011 reveló que existían 37 genes compartidos por todos los virus de la familia (core genes) [Garavaglia et al., 2012], mientras que los restantes eran típicos de género, de subconjuntos menores o únicos por especie, hasta completar una carga génica de entre 80 y 180 secuencias codificantes según la especie considerada [Ferrelli et al., 2018]. A su vez, el análisis filogenético basado en concatémeros de MSA de los core genes de los baculovirus permitió confirmar la clásica división en los cuatro géneros Alfa, Beta, Gama y Deltabaculovirus [Miele et al., 2011]. Además, se han caracterizado funcional y bioinformáticamente regiones genómicas de los baculovirus que actúan como orígenes de replicación [Miele et al., 2015; Miele et al., 2019], regiones regulatorias implicadas en el proceso de poliadenilación [Peros et al., 2020] junto con las regiones regulatorias upstream, proteínas de un interés particular como GP64 [Parsza et al., 2020] y P74 [Nugnes et al., 2021], un análisis de ortología génica con la propuesta de un nuevo core gene [Cerrudo et al., 2023], un estudio de microARN baculovirales [Motta et al., 2024]. Por otro lado, se ha realizado también análisis de viromica en Aedes aegypti de Argentina utilizando tecnología Nanopore [Ripoll et al., 2025].
En paralelo, la creciente expertise del grupo de trabajo en el área de la bioinformática le ha permitido al LIGBCM colaborar en distintos trabajos: en el análisis de datos de RNA-seq para detectar genes expresados diferencialmente en corazón ovino fetal y adulto, proponiendo nuevos biomarcadores para la regeneración cardíaca y aportando a la anotación del transcriptoma de oveja [Locatelli et al., 2020]; en la caracterización del metagenoma de Culex quinquefasciatus mediante tecnologías de NGS [Flores et al., 2022; Flores et al., 2023], determinando su composición fúngica y bacteriana; en la determinación del cambio de la microbiota en ratones frente a la desincronización circadiana crónica del ritmo de alimentación-ayuno [Trebucq et al., 2023; Crespo et al., 2026]; y evaluando estrategias de potenciación de bioplaguicidas mediante la incorporación de factores proteicos y extractos con actividades que promueven la acción de entomopatógenos [Barrera et al., 2023]. Los aportes de la bioinformática por parte del LIGBCM-AVI en estos trabajos estuvieron vinculados con el ensamblado de secuencias transcriptómicas, de amplicones de ARN ribosomal 16S e ITS (Internal Transcribed Spacer) y de secuencias genómicas obtenidas mediante NGS, la determinación de genes diferencialmente expresados, de abundancia microbiana, la caracterización y comparación de los genomas secuenciados, la identificación de secuencias aminoacídicas con funciones particulares y la caracterización estructural de proteínas.
A modo de cierre, la historia de la bioinformática refleja también la transformación de las ciencias y tecnologías de la vida durante las últimas décadas. El pasaje desde estudios centrados en genes individuales hacia el análisis masivo e integrado de genomas, transcriptomas, proteomas y otros conjuntos de datos biológicos modificó profundamente la manera de formular hipótesis, diseñar experimentos e interpretar resultados. En este contexto, la bioinformática dejó de ser una herramienta complementaria para convertirse en un componente central de la investigación biológica moderna. La trayectoria del LIGBCM-AVI acompaña y ejemplifica esta evolución, incorporando progresivamente nuevas estrategias computacionales para abordar problemas biológicos cada vez más complejos. Este proceso, no sólo permitió ampliar las capacidades analíticas del grupo, sino también consolidar una línea de trabajo interdisciplinaria en la que convergen biología molecular, virología, genética, programación, estadística y análisis de datos. Así, nuestro laboratorio pasó de caracterizar genes aislados de virus a estudiar cientos de genomas con fines funcionales y evolutivos, que no sólo generaron conocimientos originales, sino que posibilitaron la realización de intervenciones racionales con herramientas de ingeniería genética para potenciar biotecnologías. En la era de la biología sintética, otro emergente aplicado de este siglo dentro de las tecnologías de la vida, la bioinformática nos está permitiendo direccionar experimentos para minimizar o reescribir genomas virales que son la base de plataformas de expresión de proteínas heterólogas, o de sistemas de vehiculización de genes en medicina y veterinaria. En los próximos años, es esperable que las tecnologías emergentes, como los modelos de aprendizaje profundo aplicados a la predicción funcional de biomoléculas y los enfoques de biología de sistemas, planteen nuevos desafíos computacionales y conceptuales que requerirán una interacción aún más estrecha entre disciplinas experimentales y computacionales. En este escenario, la capacidad de transformar datos en conocimiento biológico relevante continuará dependiendo no solo del desarrollo de nuevas herramientas bioinformáticas, sino también de la formación de profesionales con una visión interdisciplinaria y de la consolidación de espacios colaborativos entre distintas áreas científicas. La experiencia del LIGBCM-AVI demuestra cómo la incorporación progresiva de la bioinformática potenció la investigación básica y aplicada, generando nuevas líneas de trabajo y contribuyendo a la formación de recursos humanos en un área estratégica para el desarrollo científico y tecnológico actual.
Abramson, J., Adler, J., Dunger, J., Evans, R., Green, T., Pritzel, A., Ronneberger, O., Willmore, L., Ballard, A. J., Bambrick, J., Bodenstein, S. W., Evans, D. A., Hung, C. C., O'Neill, M., Reiman, D., Tunyasuvunakool, K., Wu, Z., Žemgulytė, A., Arvaniti, E., … Jumper, J. M. (2024). Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 630(8016), 493–500. DOI: https://doi.org/10.1038/s41586-024-07487-w
Alanazi, W., Meng, D., & Pollastri, G. (2025). Advancements in one-dimensional protein structure prediction using machine learning and deep learning. Computational and structural biotechnology journal, 27, 1416–1430. DOI: https://doi.org/10.1016/j.csbj.2025.04.005
Albariño, C. G., Ghiringhelli, P. D., Posik, D. M., Lozano, M. E., Ambrosio, A. M., Sanchez, A., & Romanowski, V. (1997). Molecular characterization of attenuated Junin virus strains. The Journal of general virology, 78 ( Pt 7), 1605–1610. DOI: https://doi.org/10.1099/0022-1317-78-7-1605
Albariño, C. G., Posik, D. M., Ghiringhelli, P. D., Lozano, M. E., & Romanowski, V. (1998). Arenavirus phylogeny: a new insight. Virus genes, 16(1), 39–46. DOI: https://doi.org/10.1023/a:1007993525052
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., & Lipman, D. J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research, 25(17), 3389–3402. DOI: https://doi.org/10.1093/nar/25.17.3389
Amarapathy, S., & Tyagi, S. (2026). Multi-omics applications in health and diseases. Progress in molecular biology and translational science, 221, 43–70. DOI: https://doi.org/10.1016/bs.pmbts.2026.01.012
Bailey, T. L., & Elkan, C. (1994). Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proceedings. International Conference on Intelligent Systems for Molecular Biology, 2, 28–36.
Barrera, G. P., Belaich, M. N., Patarroyo, M. A., Villamizar, L. F., & Ghiringhelli, P. D. (2015). Evidence of recent interspecies horizontal gene transfer regarding nucleopolyhedrovirus infection of Spodoptera frugiperda. BMC genomics, 16, 1008. DOI: https://doi.org/10.1186/s12864-015-2218-5
Barrera, G. P., Molano, G. M., Beltrán, C., Patarroyo, M. A., Cerrudo, C. S., & Belaich, M. N. (2023). Transmission of Furcraea necrotic streak virus (FNSV) by Olpidium virulentus. Plant Pathology, 72(8), 1393-1405. DOI: https://doi.org/10.1111/ppa.13765
Barrera, G. P., Villamizar, L. F., Araque, G. A., Gómez, J. A., Guevara, E. J., Cerrudo, C. S., & Belaich, M. N. (2021). Natural Coinfection between Novel Species of Baculoviruses in Spodoptera ornithogalli Larvae. Viruses, 13(12), 2520. DOI: https://doi.org/10.3390/v13122520
Barrera, G. P., Villamizar, L. F., Espinel, C., Quintero, E. M., Belaich, M. N., Toloza, D. L., Ghiringhelli, P. D., & Vargas, G. (2017). Identification of Diatraea spp. (Lepidoptera: Crambidae) based on cytochrome oxidase II. PLOS one, 12(9), e0184053. DOI: https://doi.org/10.1371/journal.pone.0184053
Belaich, M. N., Buldain, D., Ghiringhelli, P. D., Hyman, B., Micieli, M. V., & Achinelly, M. F. (2015). Nucleotide sequence differentiation of Argentine isolates of the mosquito parasitic nematode Strelkovimermis spiculatus (Nematoda: Mermithidae). Journal of vector ecology : journal of the Society for Vector Ecology, 40(2), 415–418. DOI: https://doi.org/10.1111/jvec.12183
Belaich, M. N., Rodríguez, V. A., Bilen, M. F., Pilloff, M. G., Romanowski, V., Sciocco-Cap, A., & Ghiringhelli, P. D. (2006). Sequencing and characterisation of p74 gene in two isolates of Anticarsia gemmatalis MNPV. Virus genes, 32(1), 59–70. DOI: https://doi.org/10.1007/s11262-005-5846-z
Bernhofer, M., Dallago, C., Karl, T., Satagopam, V., Heinzinger, M., Littmann, M., Olenyi, T., Qiu, J., Schütze, K., Yachdav, G., Ashkenazy, H., Ben-Tal, N., Bromberg, Y., Goldberg, T., Kajan, L., O'Donoghue, S., Sander, C., Schafferhans, A., Schlessinger, A., Vriend, G., … Rost, B. (2021). PredictProtein - Predicting Protein Structure and Function for 29 Years. Nucleic acids research, 49(W1), W535–W540. DOI: https://doi.org/10.1093/nar/gkab354
Biasini, M., Bienert, S., Waterhouse, A., Arnold, K., Studer, G., Schmidt, T., Kiefer, F., Gallo Cassarino, T., Bertoni, M., Bordoli, L., & Schwede, T. (2014). SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic acids research, 42(Web Server issue), W252–W258. DOI: https://doi.org/10.1093/nar/gku340
Bilen, M. F., Pilloff, M. G., Belaich, M. N., Da Ros, V. G., Rodrigues, J. C., Ribeiro, B. M., Romanowski, V., Lozano, M. E., & Ghiringhelli, P. D. (2007). Functional and structural characterisation of AgMNPV ie1. Virus genes, 35(3), 549–562. DOI: https://doi.org/10.1007/s11262-007-0150-8
Bilofsky, H. S., Burks, C., Fickett, J. W., Goad, W. B., Lewitter, F. I., Rindone, W. P., Swindell, C. D., & Tung, C. S. (1986). The GenBank genetic sequence databank. Nucleic acids research, 14(1), 1–4. DOI: https://doi.org/10.1093/nar/14.1.1
Bindea, G., Mlecnik, B., Hackl, H., Charoentong, P., Tosolini, M., Kirilovsky, A., Fridman, W. H., Pagès, F., Trajanoski, Z., & Galon, J. (2009). ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics (Oxford, England), 25(8), 1091–1093. DOI: https://doi.org/10.1093/bioinformatics/btp101
Blum, M., Andreeva, A., Florentino, L. C., Chuguransky, S. R., Grego, T., Hobbs, E., Pinto, B. L., Orr, A., Paysan-Lafosse, T., Ponamareva, I., Salazar, G. A., Bordin, N., Bork, P., Bridge, A., Colwell, L., Gough, J., Haft, D. H., Letunic, I., Llinares-López, F., … Bateman, A. (2025). InterPro: the protein sequence classification resource in 2025. Nucleic acids research, 53(D1), D444–D456. DOI: https://doi.org/10.1093/nar/gkae1082
Carver, T., & Bleasby, A. (2003). The design of Jemboss: a graphical user interface to EMBOSS. Bioinformatics (Oxford, England), 19(14), 1837–1843. DOI: https://doi.org/10.1093/bioinformatics/btg251
Carver, T., Berriman, M., Tivey, A., Patel, C., Böhme, U., Barrell, B. G., Parkhill, J., & Rajandream, M. A. (2008). Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics (Oxford, England), 24(23), 2672–2676. DOI: https://doi.org/10.1093/bioinformatics/btn529
Cerrudo, C. S., Mengual Gómez, D. L., Gómez, D. E., & Ghiringhelli, P. D. (2015). Novel insights into the evolution and structural characterization of dyskerin using comprehensive bioinformatics analysis. Journal of proteome research, 14(2), 874–887. DOI: https://doi.org/10.1021/pr500956k
Cerrudo, C. S., Motta, L. F., Cuccovia Warlet, F. U., Lassalle, F. M., Simonin, J. A., & Belaich, M. N. (2023). Protein-Gene Orthology in Baculoviridae: An Exhaustive Analysis to Redefine the Ancestrally Common Coding Sequences. Viruses, 15(5), 1091. DOI: https://doi.org/10.3390/v15051091
Chen, S., Zhou, Y., Chen, Y., & Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics (Oxford, England), 34(17), i884–i890. DOI: https://doi.org/10.1093/bioinformatics/bty560
Crespo, M. T., Del Rio, A., Borio, C., Bilen, M., Chiesa, J. J., & Agostino, P. V. (2026). Antibiotic-induced Dysbiosis of Gut Microbiota Increases Food Motivation and Anticipatory Activity Under a Time-restricted Feeding Protocol. Journal of biological rhythms, 41(1), 81–93. DOI: https://doi.org/10.1177/07487304251359349
Crooks, G. E., Hon, G., Chandonia, J. M., & Brenner, S. E. (2004). WebLogo: a sequence logo generator. Genome research, 14(6), 1188–1190. DOI: https://doi.org/10.1101/gr.849004
Darriba, D., Taboada, G. L., Doallo, R., & Posada, D. (2011). ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics (Oxford, England), 27(8), 1164–1165. DOI: https://doi.org/10.1093/bioinformatics/btr088
Darriba, D., Taboada, G. L., Doallo, R., & Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nature methods, 9(8), 772. DOI: https://doi.org/10.1038/nmeth.2109
Delfino, C. M., Cerrudo, C. S., Biglione, M., Oubiña, J. R., Ghiringhelli, P. D., & Mathet, V. L. (2018). A comprehensive bioinformatic analysis of hepatitis D virus full-length genomes. Journal of viral hepatitis, 25(7), 860–869. DOI: https://doi.org/10.1111/jvh.12876
Devereux J, Haeberli P, Smithies O. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Research. 1984 Jan 11;12(1 Pt 1):387-95. DOI: 10.1093/nar/12.1part1.387. PMID: 6546423; PMCID: PMC321012.
Di Tommaso, P., Moretti, S., Xenarios, I., Orobitg, M., Montanyola, A., Chang, J. M., Taly, J. F., & Notredame, C. (2011). T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic acids research, 39(Web Server issue), W13–W17. DOI: https://doi.org/10.1093/nar/gkr245
Drozdetskiy, A., Cole, C., Procter, J., & Barton, G. J. (2015). JPred4: a protein secondary structure prediction server. Nucleic acids research, 43(W1), W389–W394. DOI: https://doi.org/10.1093/nar/gkv332
Edgar R. C. (2022). Muscle5: High-accuracy alignment ensembles enable unbiased assessments of sequence homology and phylogeny. Nature communications, 13(1), 6968. DOI: https://doi.org/10.1038/s41467-022-34630-w
Felsenstein, J. (1989). PHYLIP -- Phylogeny Inference Package (Version 3.2). Cladistics, 5, 164-166.
Ferrelli ML, Ghiringhelli PD, Belaich MN, Sciocco-Cap A, Romanowski V. The baculoviral genome (2011). The Viral Genome: Molecular Structure Diversity, Gene Expression Mechanisms and Host-Virus Interactions, pp 3-32 . Rijeka: INTECH.
Ferrelli, M. L., Pidre, M. L., Ghiringhelli, P. D., Torres, S., Fabre, M. L., Masson, T., Cédola, M. T., Sciocco-Cap, A., & Romanowski, V. (2018). Genomic analysis of an Argentinean isolate of Spodoptera frugiperda granulovirus reveals that various baculoviruses code for Lef-7 proteins with three F-box domains. PLOS one, 13(8), e0202598. DOI: https://doi.org/10.1371/journal.pone.0202598
Ferrelli, M. L., Salvador, R., Biedma, M. E., Berretta, M. F., Haase, S., Sciocco-Cap, A., Ghiringhelli, P. D., & Romanowski, V. (2012). Genome of Epinotia aporema granulovirus (EpapGV), a polyorganotropic fast killing betabaculovirus with a novel thymidylate kinase gene. BMC genomics, 13, 548. DOI: https://doi.org/10.1186/1471-2164-13-548
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Heger, A., Hetherington, K., Holm, L., Mistry, J., Sonnhammer, E. L., Tate, J., & Punta, M. (2014). Pfam: the protein families database. Nucleic acids research, 42(Database issue), D222–D230. DOI: https://doi.org/10.1093/nar/gkt1223
Finn, R. D., Clements, J., & Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Research, 39, W29–W37. DOI: https://doi.org/10.1093/nar/gkr367
Flores, G. A. M., Lopez, R. P., Cerrudo, C. S., Consolo, V. F., & Berón, C. M. (2022). Culex quinquefasciatus Holobiont: A Fungal Metagenomic Approach. Frontiers in fungal biology, 3, 918052. DOI: https://doi.org/10.3389/ffunb.2022.918052
Flores, G. A. M., Lopez, R. P., Cerrudo, C. S., Perotti, M. A., Consolo, V. F., & Berón, C. M. (2023). Wolbachia dominance influences the Culex quinquefasciatus microbiota. Scientific reports, 13(1), 18980. DOI: https://doi.org/10.1038/s41598-023-46067-2
Galaxy Community (2024). The Galaxy platform for accessible, reproducible, and collaborative data analyses: 2024 update. Nucleic acids research, 52(W1), W83–W94. DOI: https://doi.org/10.1093/nar/gkae410
Garavaglia, M. J., Miele, S. A., Iserte, J. A., Belaich, M. N., & Ghiringhelli, P. D. (2012). The ac53, ac78, ac101, and ac103 genes are newly discovered core genes in the family Baculoviridae. Journal of virology, 86(22), 12069–12079. DOI: https://doi.org/10.1128/JVI.01873-12
Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S. E., Wilkins, M. R., Appel, R. D. y Bairoch, A. (2005). Protein identification and analysis tools on the ExPASy server. The proteomics protocols handbook (pp. 571-607). Totowa: Humana Press.
Ge, S. X., Jung, D., & Yao, R. (2020). ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics (Oxford, England), 36(8), 2628–2629. DOI: https://doi.org/10.1093/bioinformatics/btz931
Ghiringhelli, P. D., Rivera Pomar, R. V., Lozano, M. E., Grau, O., & Romanowski, V. (1991). Molecular organization of Junin virus S RNA: complete nucleotide sequence, relationship with other members of the Arenaviridae and unusual secondary structures. The Journal of general virology, 72 ( Pt 9), 2129–2141. DOI: https://doi.org/10.1099/0022-1317-72-9-2129
Gouet, P., Robert, X., & Courcelle, E. (2003). ESPript/ENDscript: Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic acids research, 31(13), 3320–3323. DOI: https://doi.org/10.1093/nar/gkg556
Grundy, W. N., Bailey, T. L., Elkan, C. P., & Baker, M. E. (1997). Meta-MEME: motif-based hidden Markov models of protein families. Computer applications in the biosciences : CABIOS, 13(4), 397–406. DOI: https://doi.org/10.1093/bioinformatics/13.4.397
Hamm, G. H., & Cameron, G. N. (1986). The EMBL data library. Nucleic acids research, 14(1), 5–9. DOI: https://doi.org/10.1093/nar/14.1.5
Herráez A. (2006). Biomolecules in the computer: Jmol to the rescue. Biochemistry and molecular biology education, 34(4), 255–261. DOI: https://doi.org/10.1002/bmb.2006.494034042644
Hofmann, K., Bucher, P., Falquet, L., & Bairoch, A. (1999). The PROSITE database, its status in 1999. Nucleic acids research, 27(1), 215–219. DOI: https://doi.org/10.1093/nar/27.1.215
Holm L. (2022). Dali server: structural unification of protein families. Nucleic acids research, 50(W1), W210–W215. DOI: https://doi.org/10.1093/nar/gkac387
Hsieh, Y. C., Bockwoldt, M., & Heiland, I. (2025). ProTaxoVis-protein taxonomic visualisation of presence. BMC bioinformatics, 26(1), 128. DOI: https://doi.org/10.1186/s12859-025-06146-9
Ibañez-Lligoña, M., Colomer-Castell, S., González-Sánchez, A., Gregori, J., Campos, C., Garcia-Cehic, D., Andrés, C., Piñana, M., Pumarola, T., Rodríguez-Frias, F., Antón, A., & Quer, J. (2023). Bioinformatic Tools for NGS-Based Metagenomics to Improve the Clinical Diagnosis of Emerging, Re-Emerging and New Viruses. Viruses, 15(2), 587. DOI: https://doi.org/10.3390/v15020587
Ilgisonis, E. V., Pogodin, P. V., Kiseleva, O. I., Tarbeeva, S. N., & Ponomarenko, E. A. (2022). Evolution of Protein Functional Annotation: Text Mining Study. Journal of personalized medicine, 12(3), 479. DOI: https://doi.org/10.3390/jpm12030479
Jacques, F., Bolivar, P., Pietras, K., & Hammarlund, E. U. (2023). Roadmap to the study of gene and protein phylogeny and evolution-A practical guide. PLOS one, 18(2), e0279597. DOI: https://doi.org/10.1371/journal.pone.0279597
Kawabata T. (2003). MATRAS: A program for protein 3D structure comparison. Nucleic acids research, 31(13), 3367–3369. DOI: https://doi.org/10.1093/nar/gkg581
Kerrien, S., Alam-Faruque, Y., Aranda, B., Bancarz, I., Bridge, A., Derow, C., Dimmer, E., Feuermann, M., Friedrichsen, A., Huntley, R., Kohler, C., Khadake, J., Leroy, C., Liban, A., Lieftink, C., Montecchi-Palazzi, L., Orchard, S., Risse, J., Robbe, K., Roechert, B., … Hermjakob, H. (2007). IntAct--open source resource for molecular interaction data. Nucleic acids research, 35(Database issue), D561–D565. DOI: https://doi.org/10.1093/nar/gkl958
Kouza, M., Faraggi, E., Kolinski, A., & Kloczkowski, A. (2017). The GOR Method of Protein Secondary Structure Prediction and Its Application as a Protein Aggregation Prediction Tool. Methods in molecular biology (Clifton, N.J.), 1484, 7–24. DOI: https://doi.org/10.1007/978-1-4939-6406-2_2
Kulkarni, P., & Frommolt, P. (2017). Challenges in the Setup of Large-scale Next-Generation Sequencing Analysis Workflows. Computational and structural biotechnology journal, 15, 471–477. DOI: https://doi.org/10.1016/j.csbj.2017.10.001
Kuzu, O. F., Granerud, L. J. T., & Saatcioglu, F. (2025). Navigating the landscape of protein folding and proteostasis: from molecular chaperones to therapeutic innovations. Signal transduction and targeted therapy, 10(1), 358. DOI: https://doi.org/10.1038/s41392-025-02439-w
Lam, S. D., Das, S., Sillitoe, I., & Orengo, C. (2017). An overview of comparative modelling and resources dedicated to large-scale modelling of genome sequences. Acta crystallographica. Section D, Structural biology, 73(Pt 8), 628–640. DOI: https://doi.org/10.1107/S2059798317008920
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J., & Higgins, D. G. (2007). Clustal W and Clustal X version 2.0. Bioinformatics (Oxford, England), 23(21), 2947–2948. DOI: https://doi.org/10.1093/bioinformatics/btm404
Laskowski, R. A., MacArthur, M. W., Moss, D. S., & Thornton, J. M. (1993). PROCHECK: a program to check the stereochemical quality of protein structures. Applied Crystallography, 26(2), 283-291. DOI: https://doi.org/10.1107/S0021889892009944
Lechner, M., Findeiss, S., Steiner, L., Marz, M., Stadler, P. F., & Prohaska, S. J. (2011). Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC bioinformatics, 12, 124. DOI: https://doi.org/10.1186/1471-2105-12-124
Letunic, I., & Bork, P. (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic acids research, 49(W1), W293–W296. DOI: https://doi.org/10.1093/nar/gkab301
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009 Aug 15;25(16):2078-9. DOI: 10.1093/bioinformatics/btp352. Epub 2009 Jun 8. PMID: 19505943; PMCID: PMC2723002.
Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018 Sep 15;34(18):3094-3100. DOI: 10.1093/bioinformatics/bty191. PMID: 29750242; PMCID: PMC6137996.
Lo, R., & Gonçalves-Carneiro, D. (2023). Sensing nucleotide composition in virus RNA. Bioscience reports, 43(9), BSR20230372. DOI: https://doi.org/10.1042/BSR20230372
Locatelli, P., Belaich, M. N., López, A. E., Olea, F. D., Uranga Vega, M., Giménez, C. S., Simonin, J. A., Bauzá, M. D. R., Castillo, M. G., Cuniberti, L. A., Crottogini, A., Cerrudo, C. S., & Ghiringhelli, P. D. (2020). Novel insights into cardiac regeneration based on differential fetal and adult ovine heart transcriptomic analysis. American journal of physiology. Heart and circulatory physiology, 318(4), H994–H1007. DOI: https://doi.org/10.1152/ajpheart.00610.2019
Lozano, M. E., Posik, D. M., Albariño, C. G., Schujman, G., Ghiringhelli, P. D., Calderón, G., Sabattini, M., & Romanowski, V. (1997). Characterization of arenaviruses using a family-specific primer set for RT-PCR amplification and RFLP analysis. Its potential use for detection of uncharacterized arenaviruses. Virus research, 49(1), 79–89. DOI: https://doi.org/10.1016/s0168-1702(97)01458-5
MacCannell D. (2019). Platforms and Analytical Tools Used in Nucleic Acid Sequence-Based Microbial Genotyping Procedures. Microbiol Spectr, 7. DOI: https://doi.org/10.1128/microbiolspec.ame-0005-2018
Madeira, F., Madhusoodanan, N., Lee, J., Eusebi, A., Niewielska, A., Tivey, A. R. N., Lopez, R., & Butcher, S. (2024). The EMBL-EBI Job Dispatcher sequence analysis tools framework in 2024. Nucleic acids research, 52(W1), W521–W525. DOI: https://doi.org/10.1093/nar/gkae241
Majidian, S., Hadziahmetovic, A., Langschied, F., Pascarelli, S., Prieto-Baños, S., Rojas-Vargas, J., Quest for Orthologs Consortium, Braun, E. L., Dessimoz, C., Diallo, A. B., Durand, D., Fang, G., Gabaldón, T., Glover, N., Liberles, D. A., McWhite, C., Sonnhammer, E. L. L., Thomas, P. D., Ouangraoua, A., & Julca, I. (2025). Quest for Orthologs in the era of Data Deluge and AI: Challenges and Innovations in Orthology Prediction and Data Integration. Journal of molecular evolution, 93(6), 702–719. DOI: https://doi.org/10.1007/s00239-025-10272-6
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, et al (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature, 437, 376–380. DOI: https://doi.org/10.1038/nature03959
Kouza, M., Faraggi, E., Kolinski, A., & Kloczkowski, A. (2017). The GOR Method of Protein Secondary Structure Prediction and Its Application as a Protein Aggregation Prediction Tool. Methods in molecular biology (Clifton, N.J.), 1484, 7–24. DOI: https://doi.org/10.1007/978-1-4939-6406-2_2
Manzán, M. A., Lozano, M. E., Sciocco-Cap, A., Ghiringhelli, P. D., & Romanowski, V. (2002). Identification and characterization of the ecdysteroid UDP-glycosyltransferase gene of Epinotia aporema granulovirus. Virus genes, 24(2), 119–130. DOI: https://doi.org/10.1023/a:1014564331383
Marchler-Bauer, A., Zheng, C., Chitsaz, F., Derbyshire, M. K., Geer, L. Y., Geer, R. C., Gonzales, N. R., Gwadz, M., Hurwitz, D. I., Lanczycki, C. J., Lu, F., Lu, S., Marchler, G. H., Song, J. S., Thanki, N., Yamashita, R. A., Zhang, D., & Bryant, S. H. (2013). CDD: conserved domains and protein three-dimensional structure. Nucleic acids research, 41(Database issue), D348–D352. DOI: https://doi.org/10.1093/nar/gks1243
Matzura, O., & Wennborg, A. (1996). RNAdraw: an integrated program for RNA secondary structure calculation and analysis under 32-bit Microsoft Windows. Computer applications in the biosciences : CABIOS, 12(3), 247–249. DOI: https://doi.org/10.1093/bioinformatics/12.3.247
Meng, E. C., Goddard, T. D., Pettersen, E. F., Couch, G. S., Pearson, Z. J., Morris, J. H., & Ferrin, T. E. (2023). UCSF ChimeraX: Tools for structure building and analysis. Protein science: a publication of the Protein Society, 32(11), e4792. DOI: https://doi.org/10.1002/pro.4792
Mengual Gómez, D. L., Belaich, M. N., Rodríguez, V. A., & Ghiringhelli, P. D. (2010). Effects of fetal bovine serum deprivation in cell cultures on the production of Anticarsia gemmatalis multinucleopolyhedrovirus. BMC biotechnology, 10, 68. DOI: https://doi.org/10.1186/1472-6750-10-68
Mi, H., Huang, X., Muruganujan, A., Tang, H., Mills, C., Kang, D., & Thomas, P. D. (2017). PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic acids research, 45(D1), D183–D189. DOI: https://doi.org/10.1093/nar/gkw1138
Miele, S. A. B., Cerrudo, C. S., Parsza, C. N., Nugnes, M. V., Mengual Gómez, D. L., Belaich, M. N., & Ghiringhelli, P. D. (2019). Identification of Multiple Replication Stages and Origins in the Nucleopolyhedrovirus of Anticarsia gemmatalis. Viruses, 11(7), 648. DOI: https://doi.org/10.3390/v11070648
Miele, S. A., Garavaglia, M. J., Belaich, M. N., & Ghiringhelli, P. D. (2011). Baculovirus: molecular insights on their diversity and conservation. International journal of evolutionary biology, 2011, 379424. DOI: https://doi.org/10.4061/2011/379424
Miller, M. A., Pfeiffer, W. y Schwartz, T. (2010, november). Creating the CIPRES Science Gateway for inference of large phylogenetic trees. 2010 gateway computing environments workshop (GCE) (pp. 1-8). Ieee. DOI: https://doi.org/10.1109/GCE.2010.5676129
Minh, B. Q., Hahn, M. W., & Lanfear, R. (2020). New Methods to Calculate Concordance Factors for Phylogenomic Datasets. Molecular biology and evolution, 37(9), 2727–2733. DOI: https://doi.org/10.1093/molbev/msaa106
Miskiewicz, J., Sarzynska, J., & Szachniuk, M. (2021). How bioinformatics resources work with G4 RNAs. Briefings in bioinformatics, 22(3), bbaa201. DOI: https://doi.org/10.1093/bib/bbaa201
Mittal, A., Ali, S. E., & Mathews, D. H. (2024). Using the RNAstructure Software Package to Predict Conserved RNA Structures. Current protocols, 4(11), e70054. DOI: https://doi.org/10.1002/cpz1.70054
Moeckel, C., Zaravinos, A., & Georgakopoulos-Soares, I. (2023). Strand asymmetries across genomic processes. Computational and structural biotechnology journal, 21, 2036–2047. DOI: https://doi.org/10.1016/j.csbj.2023.03.007
Mostafavi, S., Ray, D., Warde-Farley, D., Grouios, C., & Morris, Q. (2008). GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome biology, 9 Suppl 1(Suppl 1), S4. DOI: https://doi.org/10.1186/gb-2008-9-s1-s4
Motta, L. F., Cerrudo, C. S., & Belaich, M. N. (2024). A Comprehensive Study of MicroRNA in Baculoviruses. International journal of molecular sciences, 25(1), 603. DOI: https://doi.org/10.3390/ijms25010603
Nguyen, N. T. T., Contreras-Moreira, B., Castro-Mondragon, J. A., Santana-Garcia, W., Ossio, R., Robles-Espinoza, C. D., Bahin, M., Collombet, S., Vincens, P., Thieffry, D., van Helden, J., Medina-Rivera, A., Thomas-Chollier, M. y RSAT (2018). RSAT 2018: regulatory sequence analysis tools 20th anniversary. Nucleic Acids Research, 46(W1), W209–W214. DOI: https://doi.org/10.1093/nar/gky317
Nielsen, H., Teufel, F., Brunak, S. y von Heijne, G. (2024). SignalP: The Evolution of a Web Server.Prediction of Secreted Proteins (pp. 331–367). New York: Springer.
Okonechnikov, K., Golosova, O., Fursov, M., & UGENE team (2012). Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics (Oxford, England), 28(8), 1166–1167. DOI: https://doi.org/10.1093/bioinformatics/bts091
Ou, Y. Y., Ho, Q. T., & Chang, H. T. (2023). Recent advances in features generation for membrane protein sequences: From multiple sequence alignment to pre-trained language models. Proteomics, 23(23-24), e2200494. DOI: https://doi.org/10.1002/pmic.202200494
Parola, A. D., Manzán, M. A., Lozano, M. E., Ghiringhelli, P. D., Sciocco-Cap, A., & Romanowski, V. (2002). Physical and genetic map of Epinotia aporema granulovirus genome. Virus genes, 25(3), 329–341. DOI: https://doi.org/10.1023/a:1020992412175
Parsza, C. N., Gómez, D. L. M., Simonin, J. A., Belaich, M. N., & Ghiringhelli, P. D. (2021). Evaluation of the Nucleopolyhedrovirus of Anticarsia gemmatalis as a Vector for Gene Therapy in Mammals. Current gene therapy, 21(2), 177–189. DOI: https://doi.org/10.2174/1566523220999201217155945
Peros, I. G., Cerrudo, C. S., Pilloff, M. G., Belaich, M. N., Lozano, M. E., & Ghiringhelli, P. D. (2020). Advances in the Bioinformatics Knowledge of mRNA Polyadenylation in Baculovirus Genes. Viruses, 12(12), 1395. DOI: https://doi.org/10.3390/v12121395
Pilloff, M. G., Bilen, M. F., Belaich, M. N., Lozano, M. E., & Ghiringhelli, P. D. (2003). Molecular cloning and sequence analysis of the Anticarsia gemmatalis multicapsid nuclear polyhedrosis virus GP64 glycoprotein. Virus genes, 26(1), 57–69. DOI: https://doi.org/10.1023/a:1022382106174
Redelings, B. D., Holmes, I., Lunter, G., Pupko, T., & Anisimova, M. (2024). Insertions and Deletions: Computational Methods, Evolutionary Dynamics, and Biological Applications. Molecular biology and evolution, 41(9), msae177. DOI: https://doi.org/10.1093/molbev/msae177
Ripoll, L., Iserte, J., Cerrudo, C. S., Presti, D., Serrat, J. H., Poma, R., Mangione, F. A. J., Micheloud, G. A., Gioria, V. V., Berrón, C. I., Zago, M. P., Borio, C., & Bilen, M. (2025). Insect-specific RNA viruses detection in Field-Caught Aedes aegypti mosquitoes from Argentina using NGS technology. PLOS neglected tropical diseases, 19(1), e0012792. DOI: https://doi.org/10.1371/journal.pntd.0012792
Rodríguez, V. A., Belaich, M. N. y Ghiringhelli, P. D. (2011a). Baculoviruses: members of Integrated Pest Management strategies. Integrated Pest Management and Pest Control (pp. 463-480). Rijeka: INTECH.
Rodríguez VA, Belaich MN, Quintana G, Sciocco-Cap A, Ghiringhelli PD. Isolation and characterization of a Nucleopolyhedrovirus from Rachiplusia nu (Guenée) (Lepidoptera: Noctuidae) (2012). International Journal of Virology and Molecular Biology. 1: 28-34. DOI: https://doi.org/10.5923j.ijvmb.20120103.02
Rodríguez, V. A., Belaich, M. N., Gómez, D. L., Sciocco-Cap, A., & Ghiringhelli, P. D. (2011b). Identification of nucleopolyhedrovirus that infect Nymphalid butterflies Agraulis vanillae and Dione juno. Journal of invertebrate pathology, 106(2), 255–262. DOI: https://doi.org/10.1016/j.jip.2010.10.008
Roy, A., Kucukural, A., & Zhang, Y. (2010). I-TASSER: a unified platform for automated protein structure and function prediction. Nature protocols, 5(4), 725–738. DOI: https://doi.org/10.1038/nprot.2010.5
Samson, S., Lord, É., & Makarenkov, V. (2022). SimPlot++: a Python application for representing sequence similarity and detecting recombination. Bioinformatics (Oxford, England), 38(11), 3118–3120. DOI: https://doi.org/10.1093/bioinformatics/btac287
Sanger, F., Nicklen, S., & Coulson, A. R. (1977). DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences of the United States of America, 74(12), 5463–5467. DOI: https://doi.org/10.1073/pnas.74.12.5463
Sato, K., & Hamada, M. (2023). Recent trends in RNA informatics: a review of machine learning and deep learning for RNA secondary structure prediction and RNA drug discovery. Briefings in bioinformatics, 24(4), bbad186. DOI: https://doi.org/10.1093/bib/bbad186
Schneider, T. D., & Stephens, R. M. (1990). Sequence logos: a new way to display consensus sequences. Nucleic acids research, 18(20), 6097–6100. DOI: https://doi.org/10.1093/nar/18.20.6097
Sciocco-Cap, A., Parola, A. D., Goldberg, A. V., Ghiringhelli, P. D., & Romanowski, V. (2001). Characterization of a granulovirus isolated from Epinotia aporema Wals. (Lepidoptera: Tortricidae) larvae. Applied and environmental microbiology, 67(8), 3702–3706. DOI: https://doi.org/10.1128/AEM.67.8.3702-3706.2001
Sherman, B. T., Hao, M., Qiu, J., Jiao, X., Baseler, M. W., Lane, H. C., Imamichi, T., & Chang, W. (2022). DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic acids research, 50(W1), W216–W221. DOI: https://doi.org/10.1093/nar/gkac194
Sigrist, C. J. A., Cuche, B. A., de Castro, E., Coudert, E., Redaschi, N., & Bridge, A. (2026). The PROSITE database for protein families, domains, and sites. Nucleic acids research, 54(D1), D451–D458. DOI: https://doi.org/10.1093/nar/gkaf1188
Simonin, J. A., Cuccovia Warlet, F. U., Bauzá, M. D. R., Plastine, M. D. P., Alfonso, V., Olea, F. D., Cerrudo, C. S., & Belaich, M. N. (2025). Early to Late VSV-G Expression in AcMNPV BV Enhances Transduction in Mammalian Cells but Does Not Affect Virion Yield in Insect Cells. Vaccines, 13(7), 693. DOI: https://doi.org/10.3390/vaccines13070693
Sims, G. E., Jun, S. R., Wu, G. A., & Kim, S. H. (2009). Whole-genome phylogeny of mammals: evolutionary information in genic and nongenic regions. Proceedings of the National Academy of Sciences of the United States of America, 106(40), 17077–17082. DOI: https://doi.org/10.1073/pnas.0909377106
Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P. L., & Ideker, T. (2011). Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics (Oxford, England), 27(3), 431–432. DOI: https://doi.org/10.1093/bioinformatics/btq675
Söding J. (2005). Protein homology detection by HMM-HMM comparison. Bioinformatics (Oxford, England), 21(7), 951–960. DOI: https://doi.org/10.1093/bioinformatics/bti125
Stanke, M., Steinkamp, R., Waack, S., & Morgenstern, B. (2004). AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic acids research, 32, W309–W312. https://doi.org/10.1093/nar/gkh379
Steinegger, M., Meier, M., Mirdita, M., Vöhringer, H., Haunsberger, S. J., & Söding, J. (2019). HH-suite3 for fast remote homology detection and deep protein annotation. BMC bioinformatics, 20(1), 473. DOI: https://doi.org/10.1186/s12859-019-3019-7
Stothard P. (2000). The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. BioTechniques, 28(6), 1102–1104. DOI: https://doi.org/10.2144/00286ir01
Szklarczyk, D., Franceschini, A., Kuhn, M., Simonovic, M., Roth, A., Minguez, P., Doerks, T., Stark, M., Muller, J., Bork, P., Jensen, L. J., & von Mering, C. (2011). The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic acids research, 39(Database issue), D561–D568. DOI: https://doi.org/10.1093/nar/gkq973
Tamura, K., Stecher, G., & Kumar, S. (2021). MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Molecular biology and evolution, 38(7), 3022–3027. DOI: https://doi.org/10.1093/molbev/msab120
Tortorici, M. A., Albariño, C. G., Posik, D. M., Ghiringhelli, P. D., Lozano, M. E., Rivera Pomar, R., & Romanowski, V. (2001a). Arenavirus nucleocapsid protein displays a transcriptional antitermination activity in vivo. Virus research, 73(1), 41–55. DOI: https://doi.org/10.1016/s0168-1702(00)00222-7
Tortorici, M. A., Ghiringhelli, P. D., Lozano, M. E., Albariño, C. G., & Romanowski, V. (2001b). Zinc-binding properties of Junín virus nucleocapsid protein. The Journal of general virology, 82(Pt 1), 121–128. DOI: https://doi.org/10.1099/0022-1317-82-1-121
Trebucq, L. L., Lamberti, M. L., Rota, R., Aiello, I., Borio, C., Bilen, M., Golombek, D. A., Plano, S. A., & Chiesa, J. J. (2023). Chronic circadian desynchronization of feeding-fasting rhythm generates alterations in daily glycemia, LDL cholesterolemia and microbiota composition in mice. Frontiers in nutrition, 10, 1154647. DOI: https://doi.org/10.3389/fnut.2023.1154647
Van Almsick, V., Sobkowiak, A., & Schwierzeck, V. (2026). Long-read sequencing for bacterial plasmid analysis: a brief overview. FEMS microbiology letters, 373, fnag014. DOI: https://doi.org/10.1093/femsle/fnag014
Wiederstein, M., & Sippl, M. J. (2007). ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic acids research, 35(Web Server issue), W407–W410. DOI: https://doi.org/10.1093/nar/gkm290
Will, S., Joshi, T., Hofacker, I. L., Stadler, P. F., & Backofen, R. (2012). LocARNA-P: accurate boundary prediction and improved detection of structural RNAs. RNA (New York, N.Y.), 18(5), 900–914. DOI: https://doi.org/10.1261/rna.029041.111
Womble, D. D. (2000). GCG: The Wisconsin Package of sequence analysis programs. Bioinformatics Methods and Protocols (pp. 3–22). Totowa: Humana Press.
Wu, S., & Zhang, Y. (2007). LOMETS: a local meta-threading-server for protein structure prediction. Nucleic acids research, 35(10), 3375–3382. DOI: https://doi.org/10.1093/nar/gkm251
Xu, D., & Zhang, Y. (2012). Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins, 80(7), 1715–1735. DOI: https://doi.org/10.1002/prot.24065
Yang, J., Wang, J., Yao, Z. J., Jin, Q., Shen, Y., & Chen, R. (2003). GenomeComp: a visualization tool for microbial genome comparison. Journal of microbiological methods, 54(3), 423–426. DOI: https://doi.org/10.1016/s0167-7012(03)00094-0
Zanzoni, A., Montecchi-Palazzi, L., Quondam, M., Ausiello, G., Helmer-Citterich, M., & Cesareni, G. (2002). MINT: a Molecular INTeraction database. FEBS letters, 513(1), 135–140. DOI: https://doi.org/10.1016/s0014-5793(01)03293-8
Zhai, Y., Zhang, Z., & Li, Z. (2025). Advances in post-processing methods for multiple sequence alignment. Frontiers in genetics, 16, 1722317. DOI: https://doi.org/10.3389/fgene.2025.1722317
Zhang, C., Wang, Q., Li, Y., Teng, A., Hu, G., Wuyun, Q., & Zheng, W. (2024). The Historical Evolution and Significance of Multiple Sequence Alignment in Molecular Structure and Function Prediction. Biomolecules, 14(12), 1531. DOI: https://doi.org/10.3390/biom14121531
Zuker M. (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic acids research, 31(13), 3406–3415. DOI: https://doi.org/10.1093/nar/gkg595